FlowCyt: A Comparative Study of Deep Learning Approaches for Multi-Class Classification in Flow Cytometry Benchmarking
2403.00024
0
0

Abstract
This paper presents FlowCyt, the first comprehensive benchmark for multi-class single-cell classification in flow cytometry data. The dataset comprises bone marrow samples from 30 patients, with each cell characterized by twelve markers. Ground truth labels identify five hematological cell types: T lymphocytes, B lymphocytes, Monocytes, Mast cells, and Hematopoietic Stem/Progenitor Cells (HSPCs). Experiments utilize supervised inductive learning and semi-supervised transductive learning on up to 1 million cells per patient. Baseline methods include Gaussian Mixture Models, XGBoost, Random Forests, Deep Neural Networks, and Graph Neural Networks (GNNs). GNNs demonstrate superior performance by exploiting spatial relationships in graph-encoded data. The benchmark allows standardized evaluation of clinically relevant classification tasks, along with exploratory analyses to gain insights into hematological cell phenotypes. This represents the first public flow cytometry benchmark with a richly annotated, heterogeneous dataset. It will empower the development and rigorous assessment of novel methodologies for single-cell analysis.
Create account to get full access
Overview
- This paper presents a comparative study of deep learning approaches for multi-class classification in flow cytometry, a technique used to analyze and characterize cells.
- The researchers evaluated the performance of different deep learning models on a benchmark dataset to determine the most effective approach for this type of cell analysis.
- The findings have implications for improving the accuracy and efficiency of flow cytometry analysis, which is widely used in fields like immunology, cancer research, and stem cell biology.
Plain English Explanation
Flow cytometry is a powerful tool that scientists use to study individual cells. It works by passing cells through a laser beam and measuring the light signals they produce. These light signals provide information about the cells' size, shape, and internal structure, allowing researchers to identify and categorize different cell types.
In this study, the researchers explored the use of deep learning, a type of artificial intelligence, to automate the process of classifying cells based on their flow cytometry data. They compared the performance of several deep learning models on a benchmark dataset, which is a standardized collection of flow cytometry data that researchers can use to test and compare different analysis methods.
The researchers found that certain deep learning approaches were more effective than others at accurately classifying the different cell types in the benchmark dataset. By identifying the most accurate and efficient deep learning models for this task, the researchers hope to help flow cytometry researchers streamline their data analysis and gain new insights into cell biology.
The findings of this study could have important implications for fields like immunology, cancer research, and stem cell biology, where flow cytometry is widely used to study and understand the behavior of different cell types.
Technical Explanation
The researchers evaluated the performance of several deep learning models for the task of multi-class classification in flow cytometry. They used a benchmark dataset called FlowCyt, which consists of flow cytometry data from various cell types, to train and test the models.
The deep learning models they tested included convolutional neural networks (CNNs), recurrent neural networks (RNNs), and transformer-based models. The researchers compared the accuracy, precision, recall, and F1-score of these models on the FlowCyt dataset, as well as their computational efficiency in terms of training and inference time.
Their results showed that the transformer-based models, such as Neural Cellular Automata, outperformed the other deep learning approaches in terms of classification accuracy and efficiency. The researchers attribute this to the transformer's ability to capture long-range dependencies in the flow cytometry data, which is crucial for accurately identifying and classifying different cell types.
The findings of this study highlight the potential of deep learning techniques, particularly transformer-based models, for automating and improving the analysis of flow cytometry data. This could lead to faster and more accurate cell classification, which could have significant impact on various biomedical applications that rely on flow cytometry.
Critical Analysis
While the study presents a comprehensive evaluation of deep learning approaches for flow cytometry classification, there are a few potential limitations and areas for further research:
-
Dataset size and diversity: The researchers used a single benchmark dataset, FlowCyt, which may not capture the full range of variability in flow cytometry data encountered in real-world settings. Expanding the evaluation to include more diverse datasets could provide a more robust assessment of the models' performance.
-
Interpretability: Deep learning models, such as transformer-based architectures, can be complex and less interpretable than traditional statistical methods. Further research is needed to improve the interpretability of these models, which is crucial for gaining insights into the underlying biological processes captured by the flow cytometry data.
-
Real-world deployment: The study focused on evaluating the models' performance on a benchmark dataset, but the practical implementation of these deep learning approaches in real-world flow cytometry workflows may face additional challenges, such as data processing, model integration, and user adoption.
-
Generalization to other tasks: The researchers' focus was on multi-class classification, but flow cytometry data can also be used for other tasks, such as cell clustering, anomaly detection, and predictive modeling. Exploring the applicability of deep learning models to these additional use cases could further expand the utility of these approaches in flow cytometry analysis.
Despite these potential limitations, the study's findings provide valuable insights into the effectiveness of deep learning for flow cytometry analysis and lay the groundwork for future research and development in this area.
Conclusion
This paper presents a comparative study of deep learning approaches for multi-class classification in flow cytometry, a widely used technique in various biomedical fields. The researchers found that transformer-based models, such as Neural Cellular Automata, outperformed other deep learning architectures in terms of classification accuracy and computational efficiency.
These findings have important implications for streamlining and improving the analysis of flow cytometry data, which is crucial for advancing our understanding of cell biology and developing new diagnostic and therapeutic tools in areas like immunology, cancer research, and stem cell biology. By automating and optimizing the classification of cells based on flow cytometry data, researchers can gain new insights more efficiently and accelerate progress in these important fields.
The study also highlights the potential of deep learning, particularly transformer-based models, to revolutionize data analysis in the life sciences. As these techniques continue to evolve and become more widely adopted, they may unlock new opportunities for discovery and innovation across a wide range of biomedical applications.
This summary was produced with help from an AI and may contain inaccuracies - check out the links to read the original source documents!
Related Papers

Injecting Hierarchical Biological Priors into Graph Neural Networks for Flow Cytometry Prediction
Fatemeh Nassajian Mojarrad, Lorenzo Bini, Thomas Matthes, St'ephane Marchand-Maillet
0
0
In the complex landscape of hematologic samples such as peripheral blood or bone marrow derived from flow cytometry (FC) data, cell-level prediction presents profound challenges. This work explores injecting hierarchical prior knowledge into graph neural networks (GNNs) for single-cell multi-class classification of tabular cellular data. By representing the data as graphs and encoding hierarchical relationships between classes, we propose our hierarchical plug-in method to be applied to several GNN models, namely, FCHC-GNN, and effectively designed to capture neighborhood information crucial for single-cell FC domain. Extensive experiments on our cohort of 19 distinct patients, demonstrate that incorporating hierarchical biological constraints boosts performance significantly across multiple metrics compared to baseline GNNs without such priors. The proposed approach highlights the importance of structured inductive biases for gaining improved generalization in complex biological prediction tasks.
5/30/2024
🧠
BloodCell-Net: A lightweight convolutional neural network for the classification of all microscopic blood cell images of the human body
Sohag Kumar Mondal, Md. Simul Hasan Talukder, Mohammad Aljaidi, Rejwan Bin Sulaiman, Md Mohiuddin Sarker Tushar, Amjad A Alsuwaylimi
0
0
Blood cell classification and counting are vital for the diagnosis of various blood-related diseases, such as anemia, leukemia, and thrombocytopenia. The manual process of blood cell classification and counting is time-consuming, prone to errors, and labor-intensive. Therefore, we have proposed a DL based automated system for blood cell classification and counting from microscopic blood smear images. We classify total of nine types of blood cells, including Erythrocyte, Erythroblast, Neutrophil, Basophil, Eosinophil, Lymphocyte, Monocyte, Immature Granulocytes, and Platelet. Several preprocessing steps like image resizing, rescaling, contrast enhancement and augmentation are utilized. To segment the blood cells from the entire microscopic images, we employed the U-Net model. This segmentation technique aids in extracting the region of interest (ROI) by removing complex and noisy background elements. Both pixel-level metrics such as accuracy, precision, and sensitivity, and object-level evaluation metrics like Intersection over Union (IOU) and Dice coefficient are considered to comprehensively evaluate the performance of the U-Net model. The segmentation model achieved impressive performance metrics, including 98.23% accuracy, 98.40% precision, 98.25% sensitivity, 95.97% Intersection over Union (IOU), and 97.92% Dice coefficient. Subsequently, a watershed algorithm is applied to the segmented images to separate overlapped blood cells and extract individual cells. We have proposed a BloodCell-Net approach incorporated with custom light weight convolutional neural network (LWCNN) for classifying individual blood cells into nine types. Comprehensive evaluation of the classifier's performance is conducted using metrics including accuracy, precision, recall, and F1 score. The classifier achieved an average accuracy of 97.10%, precision of 97.19%, recall of 97.01%, and F1 score of 97.10%.
5/27/2024
🧪
Kernel-Based Testing for Single-Cell Differential Analysis
Anthony Ozier-Lafontaine, Camille Fourneaux, Ghislain Durif, Polina Arsenteva, C'eline Vallot, Olivier Gandrillon, Sandrine Giraud, Bertrand Michel, Franck Picard
0
0
Single-cell technologies offer insights into molecular feature distributions, but comparing them poses challenges. We propose a kernel-testing framework for non-linear cell-wise distribution comparison, analyzing gene expression and epigenomic modifications. Our method allows feature-wise and global transcriptome/epigenome comparisons, revealing cell population heterogeneities. Using a classifier based on embedding variability, we identify transitions in cell states, overcoming limitations of traditional single-cell analysis. Applied to single-cell ChIP-Seq data, our approach identifies untreated breast cancer cells with an epigenomic profile resembling persister cells. This demonstrates the effectiveness of kernel testing in uncovering subtle population variations that might be missed by other methods.
4/15/2024
DinoBloom: A Foundation Model for Generalizable Cell Embeddings in Hematology
Valentin Koch, Sophia J. Wagner, Salome Kazeminia, Ece Sancar, Matthias Hehr, Julia Schnabel, Tingying Peng, Carsten Marr
0
0
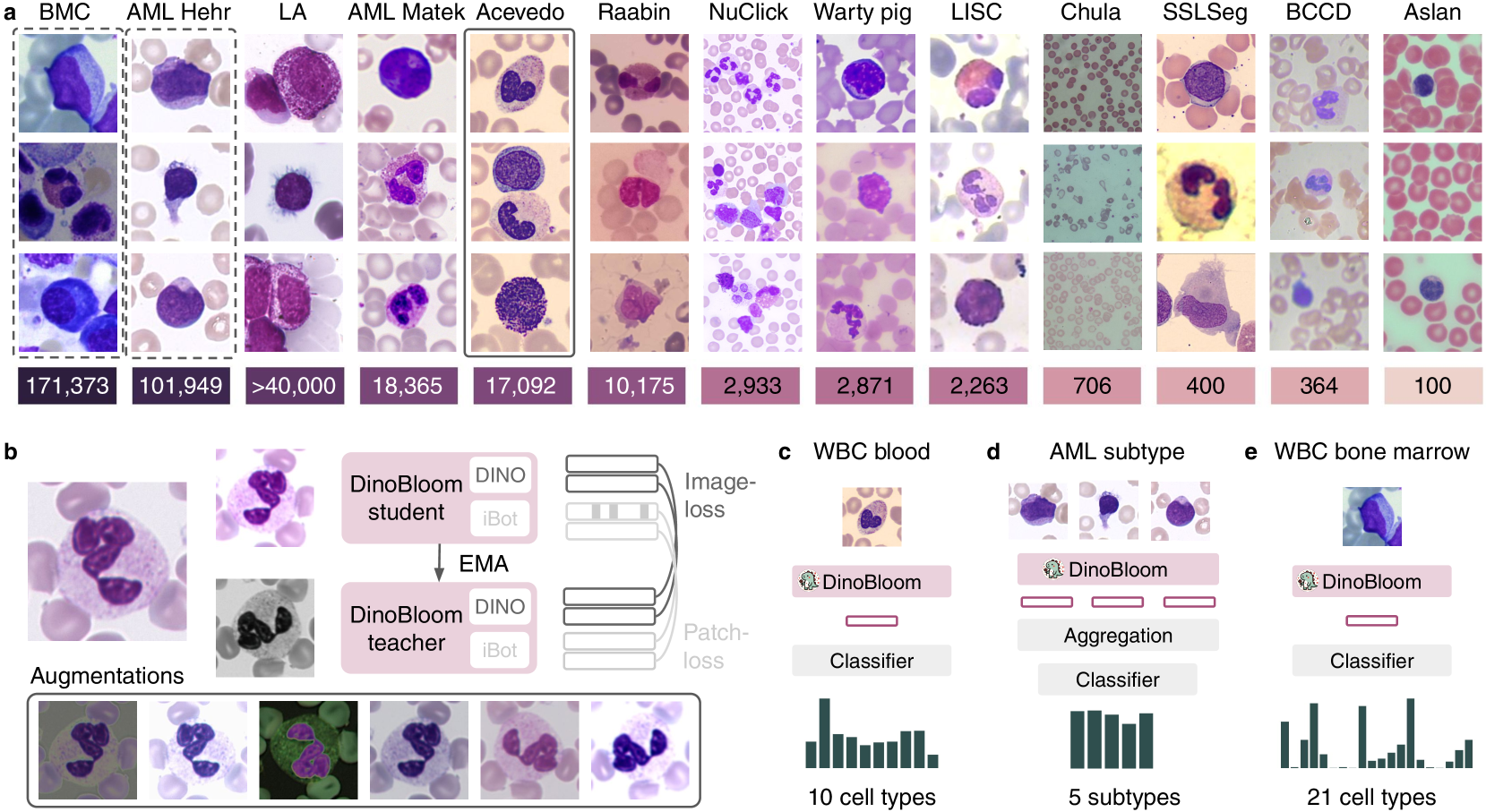
In hematology, computational models offer significant potential to improve diagnostic accuracy, streamline workflows, and reduce the tedious work of analyzing single cells in peripheral blood or bone marrow smears. However, clinical adoption of computational models has been hampered by the lack of generalization due to large batch effects, small dataset sizes, and poor performance in transfer learning from natural images. To address these challenges, we introduce DinoBloom, the first foundation model for single cell images in hematology, utilizing a tailored DINOv2 pipeline. Our model is built upon an extensive collection of 13 diverse, publicly available datasets of peripheral blood and bone marrow smears, the most substantial open-source cohort in hematology so far, comprising over 380,000 white blood cell images. To assess its generalization capability, we evaluate it on an external dataset with a challenging domain shift. We show that our model outperforms existing medical and non-medical vision models in (i) linear probing and k-nearest neighbor evaluations for cell-type classification on blood and bone marrow smears and (ii) weakly supervised multiple instance learning for acute myeloid leukemia subtyping by a large margin. A family of four DinoBloom models (small, base, large, and giant) can be adapted for a wide range of downstream applications, be a strong baseline for classification problems, and facilitate the assessment of batch effects in new datasets. All models are available at github.com/marrlab/DinoBloom.
4/9/2024