Understanding active learning of molecular docking and its applications
2406.12919
0
0

Abstract
With the advancing capabilities of computational methodologies and resources, ultra-large-scale virtual screening via molecular docking has emerged as a prominent strategy for in silico hit discovery. Given the exhaustive nature of ultra-large-scale virtual screening, active learning methodologies have garnered attention as a means to mitigate computational cost through iterative small-scale docking and machine learning model training. While the efficacy of active learning methodologies has been empirically validated in extant literature, a critical investigation remains in how surrogate models can predict docking score without considering three-dimensional structural features, such as receptor conformation and binding poses. In this paper, we thus investigate how active learning methodologies effectively predict docking scores using only 2D structures and under what circumstances they may work particularly well through benchmark studies encompassing six receptor targets. Our findings suggest that surrogate models tend to memorize structural patterns prevalent in high docking scored compounds obtained during acquisition steps. Despite this tendency, surrogate models demonstrate utility in virtual screening, as exemplified in the identification of actives from DUD-E dataset and high docking-scored compounds from EnamineReal library, a significantly larger set than the initial screening pool. Our comprehensive analysis underscores the reliability and potential applicability of active learning methodologies in virtual screening campaigns.
Create account to get full access
Overview
- This paper explores the use of active learning techniques to improve molecular docking, which is a crucial step in drug discovery and design.
- Active learning involves selectively choosing which training data to acquire, rather than using a fixed dataset, in order to maximize the model's performance with limited resources.
- The authors investigate how active learning can be applied to the prediction of binding affinities between proteins and ligands (small molecules), a key aspect of molecular docking.
Plain English Explanation
Molecular docking is the process of predicting how a small molecule (called a ligand) will bind to a protein. This is an important step in developing new drugs, as it helps researchers understand how potential drug candidates might interact with their targets in the body.
<a href="https://aimodels.fyi/papers/arxiv/improved-prediction-ligand-protein-binding-affinities-by">Accurately predicting these binding affinities</a> is challenging, as it requires complex simulations of the physical and chemical interactions between the ligand and protein.
The researchers in this paper explored using an "active learning" approach to improve these predictions. Active learning means the model gets to choose which new training data it wants to learn from, rather than being given a fixed dataset. This allows the model to focus on the most informative or difficult-to-predict examples, which can lead to better performance with less overall training data.
<a href="https://aimodels.fyi/papers/arxiv/pre-training-large-scale-generated-docking-conformations">The authors tested their active learning approach on predicting binding affinities</a>, and found that it could achieve similar accuracy to traditional methods while using much less training data. This could significantly speed up the drug discovery process by reducing the experimental work required.
Technical Explanation
The paper proposes an active learning framework for improving the prediction of protein-ligand binding affinities, a key component of molecular docking.
<a href="https://aimodels.fyi/papers/arxiv/active-learning-affinity-prediction-antibodies">The active learning approach selectively acquires new training data</a> based on the model's current performance and uncertainties, rather than using a fixed dataset. This allows the model to focus on the most informative examples and obtain high accuracy with fewer training samples.
Specifically, the authors use an uncertainty sampling strategy, where the model chooses to acquire the training examples it is most uncertain about. They implement this using a Bayesian neural network, which can output calibrated predictive uncertainties along with its binding affinity predictions.
<a href="https://aimodels.fyi/papers/arxiv/deep-learning-protein-ligand-docking-are-we">The active learning framework is evaluated on multiple protein-ligand datasets</a>, and is shown to achieve comparable performance to traditional supervised learning methods while using significantly less training data. This highlights the potential of active learning to accelerate the drug discovery process by reducing the experimental burden.
<a href="https://aimodels.fyi/papers/arxiv/physics-informed-active-learning-accelerating-quantum-chemical">The authors also discuss how the active learning approach could be further improved</a> by incorporating prior knowledge about the physics and chemistry of molecular interactions. Overall, the paper demonstrates the promise of active learning techniques for enhancing computational methods in drug discovery.
Critical Analysis
The paper presents a compelling active learning approach for improving protein-ligand binding affinity prediction, a crucial step in molecular docking and drug discovery. The authors have carefully designed their experiments and provided thorough analysis to support their claims.
One potential limitation is that the active learning framework was only evaluated on a few relatively small datasets. It would be valuable to see how it scales to larger, more diverse datasets that are more representative of real-world drug discovery challenges.
Additionally, while the authors discuss the potential benefits of incorporating physical and chemical knowledge into the active learning process, the current implementation relies solely on the model's predictive uncertainties. Exploring hybrid approaches that combine data-driven and physics-informed active learning could lead to further performance improvements.
Overall, this paper makes a strong contribution to the field of computational drug discovery by demonstrating the efficacy of active learning for enhancing binding affinity prediction. The findings encourage further research into advanced machine learning techniques that can streamline the drug development pipeline.
Conclusion
This paper presents an active learning approach for improving the prediction of protein-ligand binding affinities, a critical component of molecular docking in drug discovery. By selectively acquiring the most informative training data, the active learning model achieves comparable performance to traditional supervised learning methods while using significantly less data.
The authors' findings highlight the potential of active learning to accelerate the drug discovery process by reducing the experimental burden. Further research into incorporating physical and chemical knowledge into the active learning framework could lead to even greater improvements in binding affinity prediction and computational drug design.
This summary was produced with help from an AI and may contain inaccuracies - check out the links to read the original source documents!
Related Papers
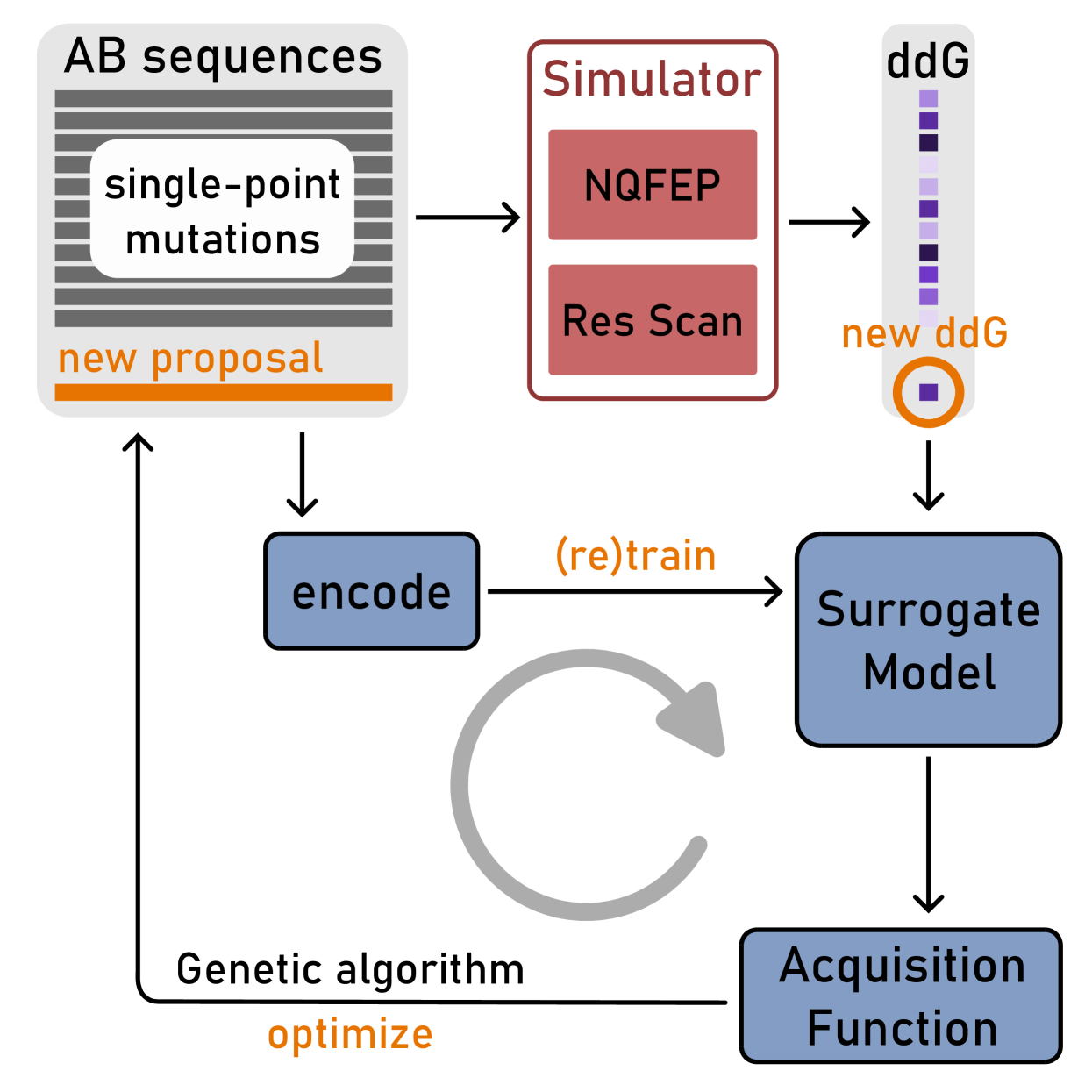
Active learning for affinity prediction of antibodies
Alexandra Gessner, Sebastian W. Ober, Owen Vickery, Dino Ogli'c, Talip Uc{c}ar
0
0
The primary objective of most lead optimization campaigns is to enhance the binding affinity of ligands. For large molecules such as antibodies, identifying mutations that enhance antibody affinity is particularly challenging due to the combinatorial explosion of potential mutations. When the structure of the antibody-antigen complex is available, relative binding free energy (RBFE) methods can offer valuable insights into how different mutations will impact the potency and selectivity of a drug candidate, thereby reducing the reliance on costly and time-consuming wet-lab experiments. However, accurately simulating the physics of large molecules is computationally intensive. We present an active learning framework that iteratively proposes promising sequences for simulators to evaluate, thereby accelerating the search for improved binders. We explore different modeling approaches to identify the most effective surrogate model for this task, and evaluate our framework both using pre-computed pools of data and in a realistic full-loop setting.
6/12/2024
🔮
Improved prediction of ligand-protein binding affinities by meta-modeling
Ho-Joon Lee, Prashant S. Emani, Mark B. Gerstein
0
0
The accurate screening of candidate drug ligands against target proteins through computational approaches is of prime interest to drug development efforts. Such virtual screening depends in part on methods to predict the binding affinity between ligands and proteins. Many computational models for binding affinity prediction have been developed, but with varying results across targets. Given that ensembling or meta-modeling methods have shown great promise in reducing model-specific biases, we develop a framework to integrate published force-field-based empirical docking and sequence-based deep learning models. In building this framework, we evaluate many combinations of individual base models, training databases, and several meta-modeling approaches. We show that many of our meta-models significantly improve affinity predictions over base models. Our best meta-models achieve comparable performance to state-of-the-art deep learning tools exclusively based on structures, while allowing for improved database scalability and flexibility through the explicit inclusion of features such as physicochemical properties or molecular descriptors. Overall, we demonstrate that diverse modeling approaches can be ensembled together to gain improvement in binding affinity prediction.
5/21/2024
🔮
Pre-Training on Large-Scale Generated Docking Conformations with HelixDock to Unlock the Potential of Protein-ligand Structure Prediction Models
Lihang Liu, Shanzhuo Zhang, Donglong He, Xianbin Ye, Jingbo Zhou, Xiaonan Zhang, Yaoyao Jiang, Weiming Diao, Hang Yin, Hua Chai, Fan Wang, Jingzhou He, Liang Zheng, Yonghui Li, Xiaomin Fang
0
0
Protein-ligand structure prediction is an essential task in drug discovery, predicting the binding interactions between small molecules (ligands) and target proteins (receptors). Recent advances have incorporated deep learning techniques to improve the accuracy of protein-ligand structure prediction. Nevertheless, the experimental validation of docking conformations remains costly, it raises concerns regarding the generalizability of these deep learning-based methods due to the limited training data. In this work, we show that by pre-training on a large-scale docking conformation generated by traditional physics-based docking tools and then fine-tuning with a limited set of experimentally validated receptor-ligand complexes, we can obtain a protein-ligand structure prediction model with outstanding performance. Specifically, this process involved the generation of 100 million docking conformations for protein-ligand pairings, an endeavor consuming roughly 1 million CPU core days. The proposed model, HelixDock, aims to acquire the physical knowledge encapsulated by the physics-based docking tools during the pre-training phase. HelixDock has been rigorously benchmarked against both physics-based and deep learning-based baselines, demonstrating its exceptional precision and robust transferability in predicting binding confirmation. In addition, our investigation reveals the scaling laws governing pre-trained protein-ligand structure prediction models, indicating a consistent enhancement in performance with increases in model parameters and the volume of pre-training data. Moreover, we applied HelixDock to several drug discovery-related tasks to validate its practical utility. HelixDock demonstrates outstanding capabilities on both cross-docking and structure-based virtual screening benchmarks.
5/24/2024
🤿
Deep Learning for Protein-Ligand Docking: Are We There Yet?
Alex Morehead, Nabin Giri, Jian Liu, Jianlin Cheng
0
0
The effects of ligand binding on protein structures and their in vivo functions carry numerous implications for modern biomedical research and biotechnology development efforts such as drug discovery. Although several deep learning (DL) methods and benchmarks designed for protein-ligand docking have recently been introduced, to date no prior works have systematically studied the behavior of docking methods within the practical context of (1) using predicted (apo) protein structures for docking (e.g., for broad applicability); (2) docking multiple ligands concurrently to a given target protein (e.g., for enzyme design); and (3) having no prior knowledge of binding pockets (e.g., for pocket generalization). To enable a deeper understanding of docking methods' real-world utility, we introduce PoseBench, the first comprehensive benchmark for practical protein-ligand docking. PoseBench enables researchers to rigorously and systematically evaluate DL docking methods for apo-to-holo protein-ligand docking and protein-ligand structure generation using both single and multi-ligand benchmark datasets, the latter of which we introduce for the first time to the DL community. Empirically, using PoseBench, we find that all recent DL docking methods but one fail to generalize to multi-ligand protein targets and also that template-based docking algorithms perform equally well or better for multi-ligand docking as recent single-ligand DL docking methods, suggesting areas of improvement for future work. Code, data, tutorials, and benchmark results are available at https://github.com/BioinfoMachineLearning/PoseBench.
6/7/2024