Advancing Extrapolative Predictions of Material Properties through Learning to Learn
2404.08657
0
0
Abstract
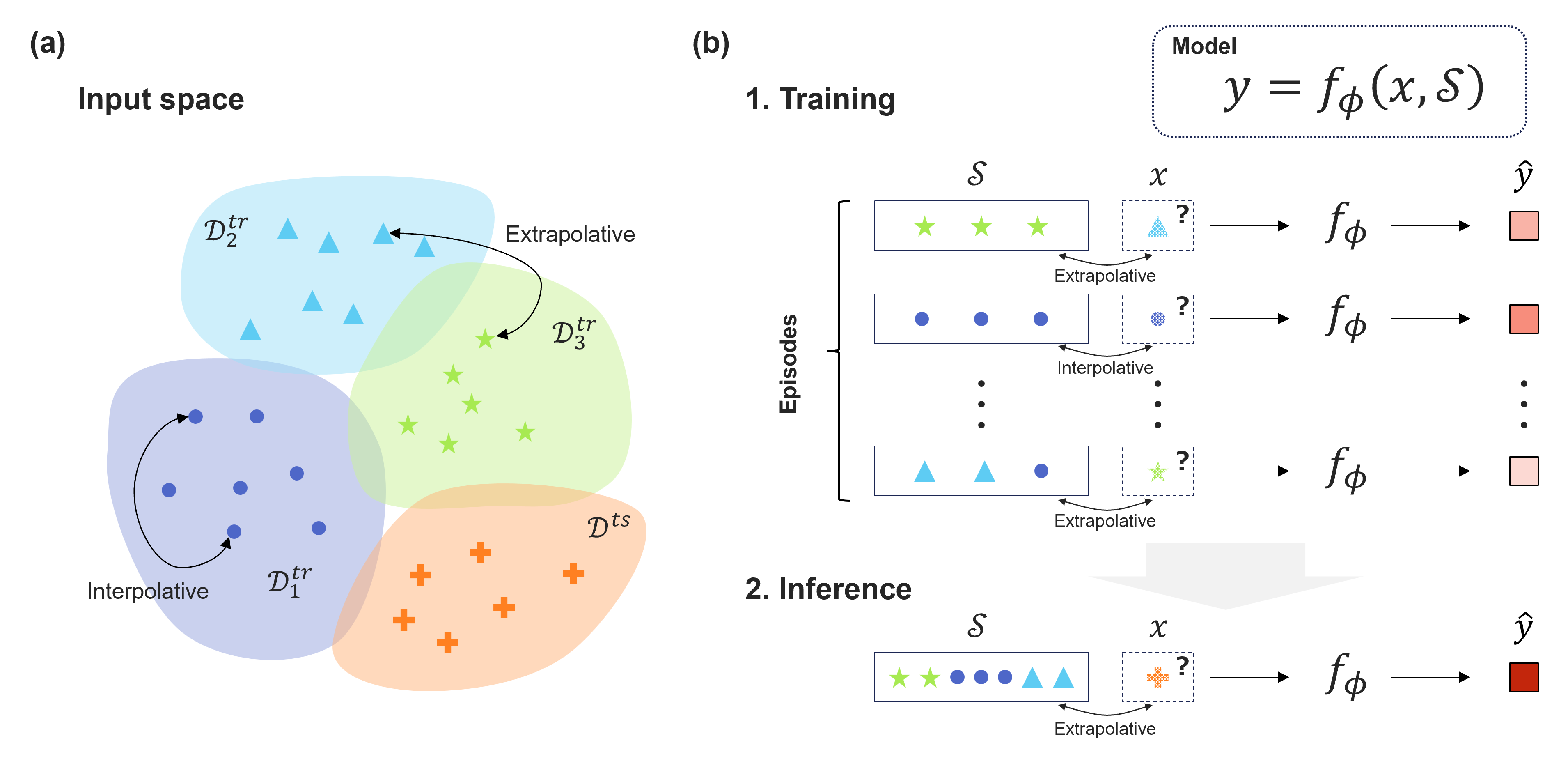
Recent advancements in machine learning have showcased its potential to significantly accelerate the discovery of new materials. Central to this progress is the development of rapidly computable property predictors, enabling the identification of novel materials with desired properties from vast material spaces. However, the limited availability of data resources poses a significant challenge in data-driven materials research, particularly hindering the exploration of innovative materials beyond the boundaries of existing data. While machine learning predictors are inherently interpolative, establishing a general methodology to create an extrapolative predictor remains a fundamental challenge, limiting the search for innovative materials beyond existing data boundaries. In this study, we leverage an attention-based architecture of neural networks and meta-learning algorithms to acquire extrapolative generalization capability. The meta-learners, experienced repeatedly with arbitrarily generated extrapolative tasks, can acquire outstanding generalization capability in unexplored material spaces. Through the tasks of predicting the physical properties of polymeric materials and hybrid organic--inorganic perovskites, we highlight the potential of such extrapolatively trained models, particularly with their ability to rapidly adapt to unseen material domains in transfer learning scenarios.
Create account to get full access
Overview
- This paper presents a new approach for improving the accuracy of predictions of material properties by leveraging meta-learning techniques.
- The proposed method, called "Learning to Learn" (L2L), aims to enable models to better extrapolate to unseen materials beyond the training data.
- The researchers demonstrate the effectiveness of L2L on several materials science tasks, including predicting the band gap energy and formation energy of new compounds.
Plain English Explanation
The paper tackles the challenge of making accurate predictions about the properties of new materials. This is important for accelerating the discovery and development of advanced materials with useful characteristics, like high-efficiency solar cells or strong, lightweight structural components.
Existing machine learning models for materials property prediction often struggle to generalize well to materials that are very different from those in the training data. The "Learning to Learn" (L2L) approach developed in this paper tries to address this limitation by training the model to learn faster and more effectively from limited data.
The key idea is to have the model learn
The researchers demonstrate that this L2L method outperforms standard machine learning techniques on several materials science benchmarks, including predicting a material's <a href="https://aimodels.fyi/papers/arxiv/machine-learning-without-processor-emergent-learning-nonlinear">band gap energy</a> and formation energy. This suggests the L2L approach could be a powerful tool for <a href="https://aimodels.fyi/papers/arxiv/active-causal-learning-decoding-chemical-complexities-targeted">accelerating the discovery and development of new functional materials</a>.
Technical Explanation
The paper introduces a "Learning to Learn" (L2L) framework for improving the extrapolation capabilities of machine learning models in materials science applications. The key innovation is training the model to learn efficiently from limited data, rather than just memorizing material-property associations.
The L2L approach involves two main components: a learning-to-learn meta-model and a base learner model. The meta-model is trained to optimize the parameters of the base learner, which is then used to make predictions on new materials. This meta-learning strategy allows the system to rapidly adapt to unseen materials by leveraging connections between different material properties.
The researchers evaluate the L2L framework on several materials science benchmarks, including predicting the band gap energy and formation energy of new compounds. They show that L2L outperforms standard supervised learning techniques, particularly when extrapolating to materials outside the training distribution.
The improvements offered by L2L are attributed to its ability to <a href="https://aimodels.fyi/papers/arxiv/mining-experimental-data-from-materials-science-literature">exploit the underlying structure and relationships in materials science data</a>. By learning how to learn efficiently, the L2L model can generalize better to new materials compared to models that simply memorize specific material-property associations.
Critical Analysis
The paper presents a promising approach for improving the extrapolation capabilities of machine learning models in materials science. The "Learning to Learn" framework is a conceptually elegant solution to the challenge of applying these models to the discovery of new materials.
However, the paper does not fully address some potential limitations of the L2L method. For example, the researchers only evaluate the approach on a relatively small number of materials science benchmarks. <a href="https://aimodels.fyi/papers/arxiv/learning-quantum-properties-from-short-range-correlations">Further testing on a wider range of materials data, including more complex systems, would be necessary to fully assess the generalizability and scalability of the L2L technique.</a>
Additionally, the paper does not provide a detailed analysis of the computational complexity and training time requirements of the L2L framework compared to standard supervised learning approaches. This information would be valuable for understanding the practical feasibility of deploying L2L in real-world materials discovery workflows.
Overall, the paper presents an innovative and promising direction for improving the predictive capabilities of machine learning in materials science. However, more research is needed to fully understand the strengths, weaknesses, and practical limitations of the L2L approach.
Conclusion
This paper introduces a new "Learning to Learn" (L2L) framework for enhancing the extrapolation abilities of machine learning models in materials science applications. The key idea is to train the model to learn efficiently from limited data, rather than just memorizing material-property associations.
The researchers demonstrate that the L2L approach outperforms standard supervised learning techniques on several materials science benchmarks, particularly when predicting the properties of new compounds outside the training distribution. This suggests that L2L could be a powerful tool for accelerating the discovery and development of advanced functional materials with desirable characteristics.
While the paper presents a conceptually elegant solution, further research is needed to fully assess the generalizability, scalability, and practical feasibility of the L2L framework. Nonetheless, this work represents an important step forward in enhancing the extrapolation capabilities of machine learning in materials science, with potentially significant implications for the field.
This summary was produced with help from an AI and may contain inaccuracies - check out the links to read the original source documents!
Related Papers

Towards robust prediction of material properties for nuclear reactor design under scarce data -- a study in creep rupture property
Yu Chen, Edoardo Patelli, Zhen Yang, Adolphus Lye
0
0
Advances in Deep Learning bring further investigation into credibility and robustness, especially for safety-critical engineering applications such as the nuclear industry. The key challenges include the availability of data set (often scarce and sparse) and insufficient consideration of the uncertainty in the data, model, and prediction. This paper therefore presents a meta-learning based approach that is both uncertainty- and prior knowledge-informed, aiming at trustful predictions of material properties for the nuclear reactor design. It is suited for robust learning under limited data. Uncertainty has been accounted for where a distribution of predictor functions are produced for extrapolation. Results suggest it achieves superior performance than existing empirical methods in rupture life prediction, a case which is typically under a small data regime. While demonstrated herein with rupture properties, this learning approach is transferable to solve similar problems of data scarcity across the nuclear industry. It is of great importance to boosting the AI analytics in the nuclear industry by proving the applicability and robustness while providing tools that can be trusted.
5/29/2024
Multimodal Learning for Materials
Viggo Moro, Charlotte Loh, Rumen Dangovski, Ali Ghorashi, Andrew Ma, Zhuo Chen, Samuel Kim, Peter Y. Lu, Thomas Christensen, Marin Soljav{c}i'c
0
0
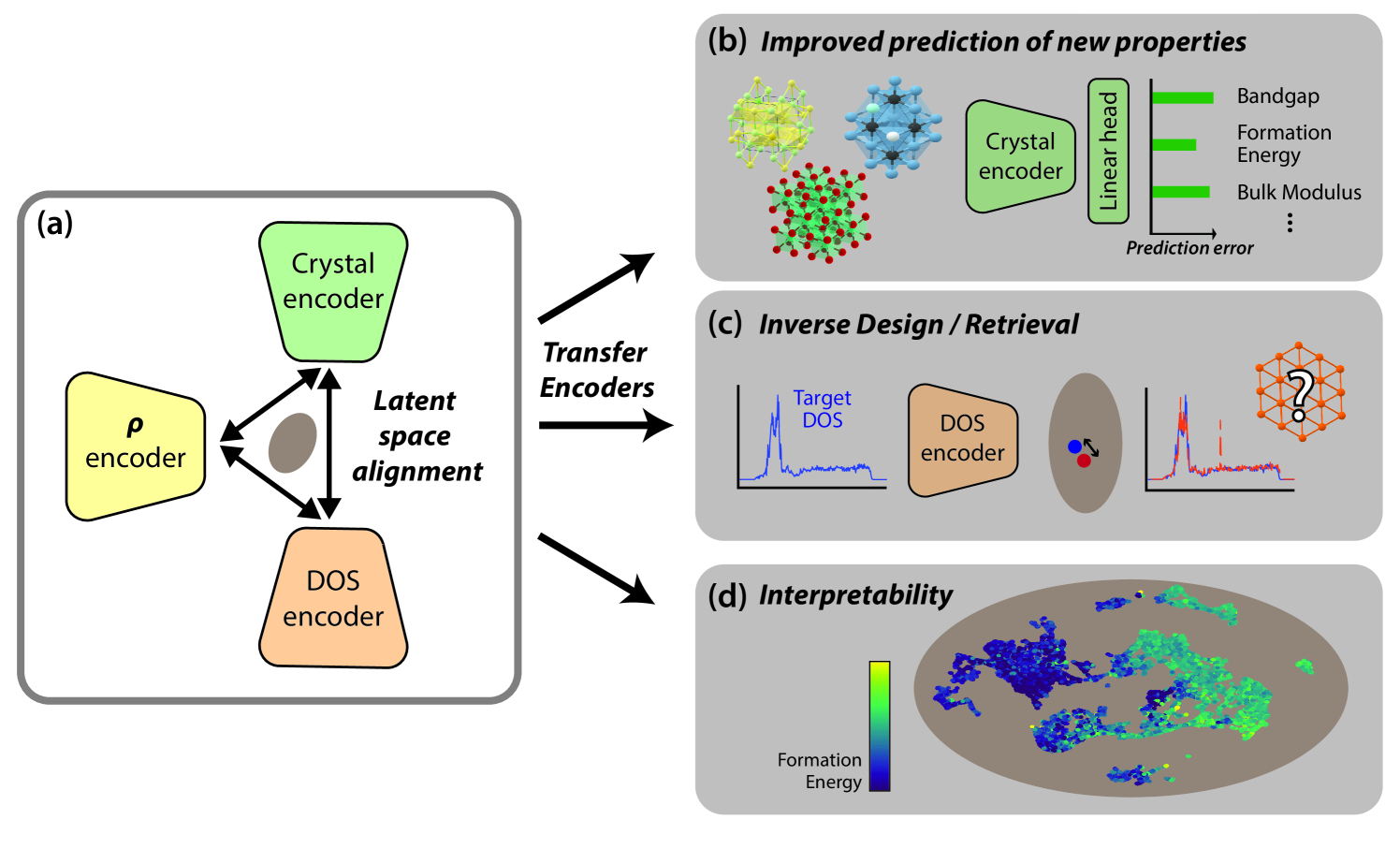
Artificial intelligence is transforming computational materials science, improving the prediction of material properties, and accelerating the discovery of novel materials. Recently, publicly available material data repositories have grown rapidly. This growth encompasses not only more materials, but also a greater variety and quantity of their associated properties. Existing machine learning efforts in materials science focus primarily on single-modality tasks, i.e., relationships between materials and a single physical property, thus not taking advantage of the rich and multimodal set of material properties. Here, we introduce Multimodal Learning for Materials (MultiMat), which enables self-supervised multi-modality training of foundation models for materials. We demonstrate our framework's potential using data from the Materials Project database on multiple axes: (i) MultiMat achieves state-of-the-art performance for challenging material property prediction tasks; (ii) MultiMat enables novel and accurate material discovery via latent space similarity, enabling screening for stable materials with desired properties; and (iii) MultiMat encodes interpretable emergent features that may provide novel scientific insights.
4/15/2024
🧠
Hybrid Quantum Graph Neural Network for Molecular Property Prediction
Michael Vitz, Hamed Mohammadbagherpoor, Samarth Sandeep, Andrew Vlasic, Richard Padbury, Anh Pham
0
0
To accelerate the process of materials design, materials science has increasingly used data driven techniques to extract information from collected data. Specially, machine learning (ML) algorithms, which span the ML discipline, have demonstrated ability to predict various properties of materials with the level of accuracy similar to explicit calculation of quantum mechanical theories, but with significantly reduced run time and computational resources. Within ML, graph neural networks have emerged as an important algorithm within the field of machine learning, since they are capable of predicting accurately a wide range of important physical, chemical and electronic properties due to their higher learning ability based on the graph representation of material and molecular descriptors through the aggregation of information embedded within the graph. In parallel with the development of state of the art classical machine learning applications, the fusion of quantum computing and machine learning have created a new paradigm where classical machine learning model can be augmented with quantum layers which are able to encode high dimensional data more efficiently. Leveraging the structure of existing algorithms, we developed a unique and novel gradient free hybrid quantum classical convoluted graph neural network (HyQCGNN) to predict formation energies of perovskite materials. The performance of our hybrid statistical model is competitive with the results obtained purely from a classical convoluted graph neural network, and other classical machine learning algorithms, such as XGBoost. Consequently, our study suggests a new pathway to explore how quantum feature encoding and parametric quantum circuits can yield drastic improvements of complex ML algorithm like graph neural network.
5/9/2024
🏅
Materials Discovery with Extreme Properties via Reinforcement Learning-Guided Combinatorial Chemistry
Hyunseung Kim (Seoul National University), Haeyeon Choi (Ewha Womans University), Dongju Kang (Seoul National University), Won Bo Lee (Seoul National University), Jonggeol Na (Ewha Womans University)
0
0
The goal of most materials discovery is to discover materials that are superior to those currently known. Fundamentally, this is close to extrapolation, which is a weak point for most machine learning models that learn the probability distribution of data. Herein, we develop reinforcement learning-guided combinatorial chemistry, which is a rule-based molecular designer driven by trained policy for selecting subsequent molecular fragments to get a target molecule. Since our model has the potential to generate all possible molecular structures that can be obtained from combinations of molecular fragments, unknown molecules with superior properties can be discovered. We theoretically and empirically demonstrate that our model is more suitable for discovering better compounds than probability distribution-learning models. In an experiment aimed at discovering molecules that hit seven extreme target properties, our model discovered 1,315 of all target-hitting molecules and 7,629 of five target-hitting molecules out of 100,000 trials, whereas the probability distribution-learning models failed. Moreover, it has been confirmed that every molecule generated under the binding rules of molecular fragments is 100% chemically valid. To illustrate the performance in actual problems, we also demonstrate that our models work well on two practical applications: discovering protein docking molecules and HIV inhibitors.
5/8/2024