AlphaCrystal-II: Distance matrix based crystal structure prediction using deep learning
2404.04810
0
0

Abstract
Computational prediction of stable crystal structures has a profound impact on the large-scale discovery of novel functional materials. However, predicting the crystal structure solely from a material's composition or formula is a promising yet challenging task, as traditional ab initio crystal structure prediction (CSP) methods rely on time-consuming global searches and first-principles free energy calculations. Inspired by the recent success of deep learning approaches in protein structure prediction, which utilize pairwise amino acid interactions to describe 3D structures, we present AlphaCrystal-II, a novel knowledge-based solution that exploits the abundant inter-atomic interaction patterns found in existing known crystal structures. AlphaCrystal-II predicts the atomic distance matrix of a target crystal material and employs this matrix to reconstruct its 3D crystal structure. By leveraging the wealth of inter-atomic relationships of known crystal structures, our approach demonstrates remarkable effectiveness and reliability in structure prediction through comprehensive experiments. This work highlights the potential of data-driven methods in accelerating the discovery and design of new materials with tailored properties.
Create account to get full access
Overview
- Presents a deep learning-based approach called AlphaCrystal-II for predicting crystal structures from only the chemical composition
- Leverages a distance matrix representation of atomic positions to learn crystal structure patterns
- Enables accurate prediction of diverse crystal structures without requiring explicit knowledge of atomic positions
Plain English Explanation
AlphaCrystal-II is a deep learning model that can predict the crystal structure of a material based only on its chemical composition. Rather than trying to directly model the 3D arrangement of atoms, the model uses a 2D distance matrix to capture the relationships between atoms.
This distance matrix representation allows the model to learn patterns in how atoms pack together to form different crystal structures, without needing to know the exact positions of the atoms ahead of time. By training on a large dataset of known crystal structures, the model can then make accurate predictions for new materials.
The advantage of this approach is that it can handle a wide variety of crystal structures, including complex ones, without requiring explicit rules or knowledge about the atomic-level details. This makes it a powerful tool for accelerating the discovery of new functional materials, which often have intricate crystal structures that are difficult to predict.
Technical Explanation
The key innovation in AlphaCrystal-II is the use of a distance matrix as the input representation for the deep learning model. Rather than directly modeling the 3D coordinates of atoms, the model takes as input a 2D matrix that encodes the distances between all pairs of atoms in the material.
This distance matrix capturing the atom-level optical chemical structure allows the model to learn patterns in how atoms pack together without needing to know the exact positions ahead of time. The model architecture then consists of convolutional and attention-based layers that can extract relevant features from this distance matrix to predict the final crystal structure.
By training on a large dataset of known crystal structures, the model is able to actively learn the relationships between atomic composition and the resulting crystal geometry. This allows it to make accurate predictions for new materials, including those with complex space group-constrained crystal structures.
Critical Analysis
The main strength of the AlphaCrystal-II approach is its ability to handle a wide variety of crystal structures without requiring explicit rules or knowledge about atomic positions. This makes it a very general and flexible tool for materials discovery.
However, a potential limitation is that the distance matrix representation may lose some information about the 3D spatial arrangement of atoms compared to directly modeling the atomic coordinates. This could make it harder for the model to capture certain structural nuances.
Additionally, the model was trained on a finite dataset of known crystal structures, so its performance may be limited for predicting completely novel or exotic crystal types that are not well-represented in the training data. Further research would be needed to assess the model's generalization capabilities.
Overall, AlphaCrystal-II represents an interesting and promising advance in the field of learning quantum properties from short-range correlations. Its distance matrix-based approach offers a unique perspective on the crystal structure prediction problem that could lead to new discoveries in materials science.
Conclusion
The AlphaCrystal-II model provides a powerful deep learning-based approach for predicting the crystal structures of materials from only their chemical composition. By using a distance matrix representation, the model can learn patterns in how atoms pack together without requiring explicit knowledge of atomic positions.
This makes AlphaCrystal-II a flexible tool for accelerating the discovery of new functional materials, which often have complex crystal structures that are difficult to predict using traditional methods. While the model has some limitations, it represents an exciting advance in the field of computational materials science and could help unlock new opportunities for innovation.
This summary was produced with help from an AI and may contain inaccuracies - check out the links to read the original source documents!
Related Papers
🔮
End-to-End Crystal Structure Prediction from Powder X-Ray Diffraction
Qingsi Lai, Lin Yao, Zhifeng Gao, Siyuan Liu, Hongshuai Wang, Shuqi Lu, Di He, Liwei Wang, Cheng Wang, Guolin Ke
0
0
Crystal structure prediction (CSP) has made significant progress, but most methods focus on unconditional generations of inorganic crystal with limited atoms in the unit cell. This study introduces XtalNet, the first equivariant deep generative model for end-to-end CSP from Powder X-ray Diffraction (PXRD). Unlike previous methods that rely solely on composition, XtalNet leverages PXRD as an additional condition, eliminating ambiguity and enabling the generation of complex organic structures with up to 400 atoms in the unit cell. XtalNet comprises two modules: a Contrastive PXRD-Crystal Pretraining (CPCP) module that aligns PXRD space with crystal structure space, and a Conditional Crystal Structure Generation (CCSG) module that generates candidate crystal structures conditioned on PXRD patterns. Evaluation on two MOF datasets (hMOF-100 and hMOF-400) demonstrates XtalNet's effectiveness. XtalNet achieves a top-10 Match Rate of 90.2% and 79% for hMOF-100 and hMOF-400 datasets in conditional crystal structure prediction task, respectively. XtalNet represents a significant advance in CSP, enabling the prediction of complex structures from PXRD data without the need for external databases or manual intervention. It has the potential to revolutionize PXRD analysis. It enables the direct prediction of crystal structures from experimental measurements, eliminating the need for manual intervention and external databases. This opens up new possibilities for automated crystal structure determination and the accelerated discovery of novel materials.
4/3/2024

Predicting Many Properties of Crystals by a Single Deep Learning Model
Haosheng Xu, Dongheng Qian, Jing Wang
0
0
The use of machine learning methods for predicting the properties of crystalline materials encounters significant challenges, primarily related to input encoding, output versatility, and interpretability. Here, we introduce CrystalBERT, an adaptable transformer-based framework with novel structure that integrates space group, elemental, and unit cell information. The method's adaptability lies not only in its ability to seamlessly combine diverse features but also in its capability to accurately predict a wide range of physically important properties, including topological properties, superconducting transition temperatures, dielectric constants, and more. CrystalBERT also provides insightful physical interpretations regarding the features that most significantly influence the target properties. Our findings indicate that space group and elemental information are more important for predicting topological and superconducting properties, in contrast to some properties that primarily depend on the unit cell information. This underscores the intricate nature of topological and superconducting properties. By incorporating all these features, we achieve a high accuracy of 91% in topological classification, surpassing prior studies and identifying previously misclassified topological materials, further demonstrating the effectiveness of our model.
5/30/2024

Diffusion Models Are Promising for Ab Initio Structure Solutions from Nanocrystalline Powder Diffraction Data
Gabe Guo, Tristan Saidi, Maxwell Terban, Simon JL Billinge, Hod Lipson
0
0
A major challenge in materials science is the determination of the structure of nanometer sized objects. Here we present a novel approach that uses a generative machine learning model based on a Diffusion model that is trained on 45,229 known structures. The model factors both the measured diffraction pattern as well as relevant statistical priors on the unit cell of atomic cluster structures. Conditioned only on the chemical formula and the information-scarce finite-size broadened powder diffraction pattern, we find that our model, PXRDnet, can successfully solve simulated nanocrystals as small as 10 angstroms across 200 materials of varying symmetry and complexity, including structures from all seven crystal systems. We show that our model can determine structural solutions with up to $81.5%$ accuracy, as measured by structural correlation. Furthermore, PXRDnet is capable of solving structures from noisy diffraction patterns gathered in real-world experiments. We suggest that data driven approaches, bootstrapped from theoretical simulation, will ultimately provide a path towards determining the structure of previously unsolved nano-materials.
6/18/2024
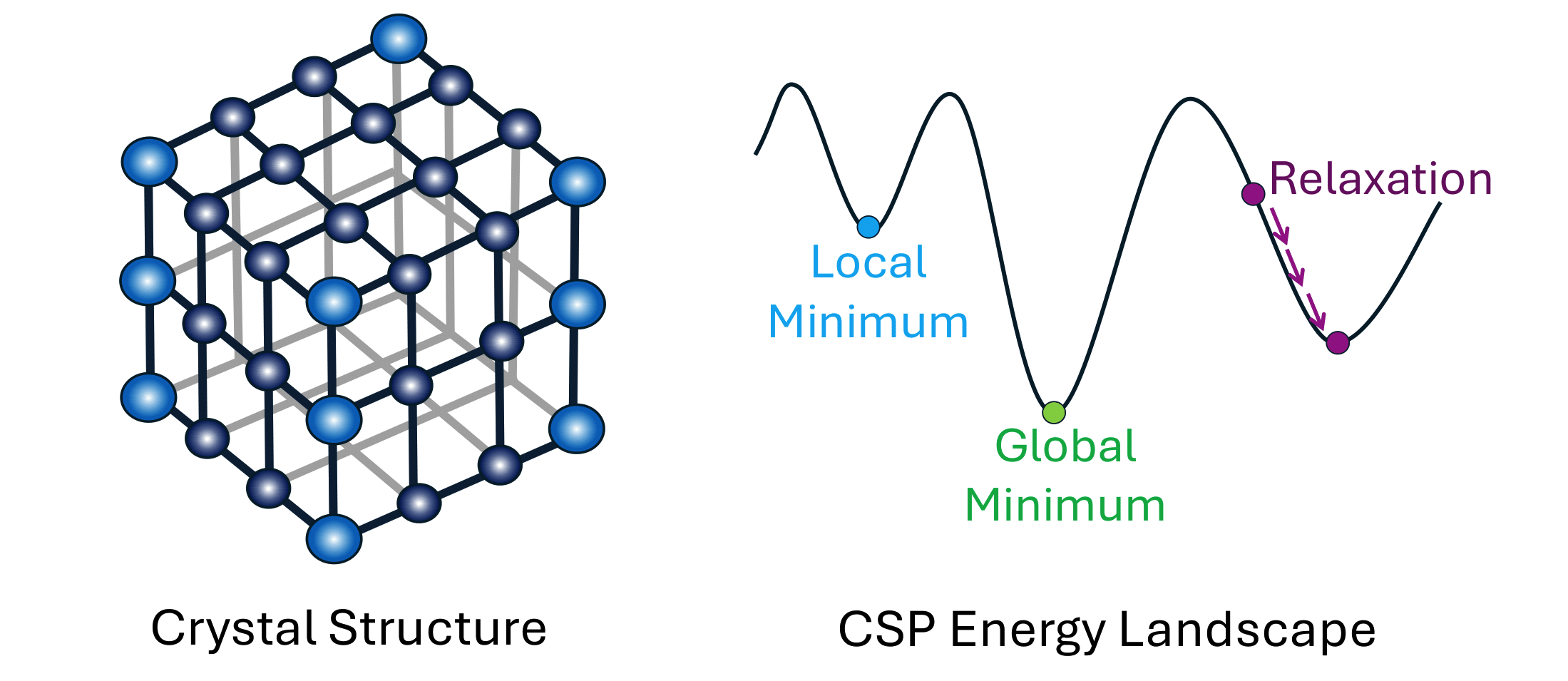
Multi-Objective Quality-Diversity for Crystal Structure Prediction
Hannah Janmohamed, Marta Wolinska, Shikha Surana, Thomas Pierrot, Aron Walsh, Antoine Cully
0
0
Crystal structures are indispensable across various domains, from batteries to solar cells, and extensive research has been dedicated to predicting their properties based on their atomic configurations. However, prevailing Crystal Structure Prediction methods focus on identifying the most stable solutions that lie at the global minimum of the energy function. This approach overlooks other potentially interesting materials that lie in neighbouring local minima and have different material properties such as conductivity or resistance to deformation. By contrast, Quality-Diversity algorithms provide a promising avenue for Crystal Structure Prediction as they aim to find a collection of high-performing solutions that have diverse characteristics. However, it may also be valuable to optimise for the stability of crystal structures alongside other objectives such as magnetism or thermoelectric efficiency. Therefore, in this work, we harness the power of Multi-Objective Quality-Diversity algorithms in order to find crystal structures which have diverse features and achieve different trade-offs of objectives. We analyse our approach on 5 crystal systems and demonstrate that it is not only able to re-discover known real-life structures, but also find promising new ones. Moreover, we propose a method for illuminating the objective space to gain an understanding of what trade-offs can be achieved.
6/24/2024