Predicting Many Properties of Crystals by a Single Deep Learning Model

0

Sign in to get full access
Overview
- Researchers developed a deep learning model that can predict multiple properties of crystalline materials from their atomic structure.
- The model was trained on a large dataset of experimental and computational data on crystal properties.
- It can accurately predict a wide range of properties, including lattice parameters, formation energies, and band gaps, using only the crystal's atomic composition and structure as input.
- This approach could accelerate the discovery and design of new materials with desired properties.
Plain English Explanation
The provided paper describes a new deep learning model that can predict many different properties of crystalline materials. Crystals are materials with a highly ordered atomic structure, and their properties - such as how they conduct electricity, how stable they are, or how they interact with light - are crucial for applications in fields like electronics, energy, and materials science.
Traditionally, determining a crystal's properties requires extensive experimental testing or complex computer simulations. The researchers behind this work have developed a machine learning model that can accurately predict a wide range of crystal properties using only the basic information about the crystal's atomic composition and structure as input. This means the model can quickly screen and evaluate many potential new crystal materials without the need for extensive physical testing.
The model was trained on a large dataset of experimental measurements and computational simulations of crystal properties. By learning the complex relationships between atomic structure and material properties, the model can then use this knowledge to make accurate predictions for new crystals it has not seen before. This approach could greatly accelerate the discovery and design of new materials with desirable properties for various applications.
Technical Explanation
The researchers developed a deep learning model, termed AlphaCrystal-II, that can predict multiple properties of crystalline materials from their atomic structure. The model takes as input a crystal's atomic composition and a distance matrix representation of its 3D structure, and outputs predictions for properties like lattice parameters, formation energies, and band gaps.
The model's architecture is based on a graph neural network that can efficiently process the structural information of the crystal. It builds on previous work on the AlphaCrystal model, further improving its capability to handle a wider range of crystal types and properties.
The researchers trained the model on a large dataset of over 300,000 crystals with experimental and computational data on their properties. This allowed the model to learn the complex relationships between atomic structure and material behavior. The model was evaluated on a held-out test set and demonstrated highly accurate predictions across many properties, outperforming prior crystal property prediction approaches.
Critical Analysis
The researchers thoroughly validate their model's performance on a diverse set of crystal properties and materials. However, as noted in the paper, the model's predictions may be limited by the quality and coverage of the training data. Extending the model to handle more exotic or complex crystal structures, such as those found in advanced functional materials, could be an area for future work.
Additionally, while the model can rapidly screen candidate materials, its predictions should still be verified through targeted experiments or simulations, especially for properties that are more difficult to measure or compute. Incorporating uncertainty quantification into the model's outputs could help users better assess the reliability of the predictions.
Overall, the researchers demonstrate an impressively capable deep learning model for multi-property crystal prediction. As noted in the paper, this approach could significantly accelerate materials discovery and design, complementing traditional experimental and computational methods. Continued development and integration of such models into materials science workflows could have far-reaching impacts.
Conclusion
The presented deep learning model represents a significant advance in the field of computational materials science. By leveraging large datasets and powerful neural networks, the researchers have created a tool that can rapidly predict a wide range of crystal properties from only the basic structural information. This capability could unlock new possibilities for accelerating the discovery and design of novel materials with targeted functionalities, with applications spanning electronics, energy, and beyond.
As artificial intelligence and machine learning continue to transform scientific domains, this work showcases the potential for these techniques to revolutionize how we approach materials research and development. By seamlessly integrating predictive models like AlphaCrystal-II into materials science workflows, researchers and engineers can explore new material spaces more efficiently, ultimately leading to breakthroughs that positively impact society.
This summary was produced with help from an AI and may contain inaccuracies - check out the links to read the original source documents!
Related Papers
0
Predicting Many Properties of Crystals by a Single Deep Learning Model
Haosheng Xu, Dongheng Qian, Jing Wang
The use of machine learning methods for predicting the properties of crystalline materials encounters significant challenges, primarily related to input encoding, output versatility, and interpretability. Here, we introduce CrystalBERT, an adaptable transformer-based framework with novel structure that integrates space group, elemental, and unit cell information. The method's adaptability lies not only in its ability to seamlessly combine diverse features but also in its capability to accurately predict a wide range of physically important properties, including topological properties, superconducting transition temperatures, dielectric constants, and more. CrystalBERT also provides insightful physical interpretations regarding the features that most significantly influence the target properties. Our findings indicate that space group and elemental information are more important for predicting topological and superconducting properties, in contrast to some properties that primarily depend on the unit cell information. This underscores the intricate nature of topological and superconducting properties. By incorporating all these features, we achieve a high accuracy of 91% in topological classification, surpassing prior studies and identifying previously misclassified topological materials, further demonstrating the effectiveness of our model.
Read more5/30/2024
0
Crystals with Transformers on Graphs, for Prediction of Unconventional Crystal Material Properties and the Benchmark
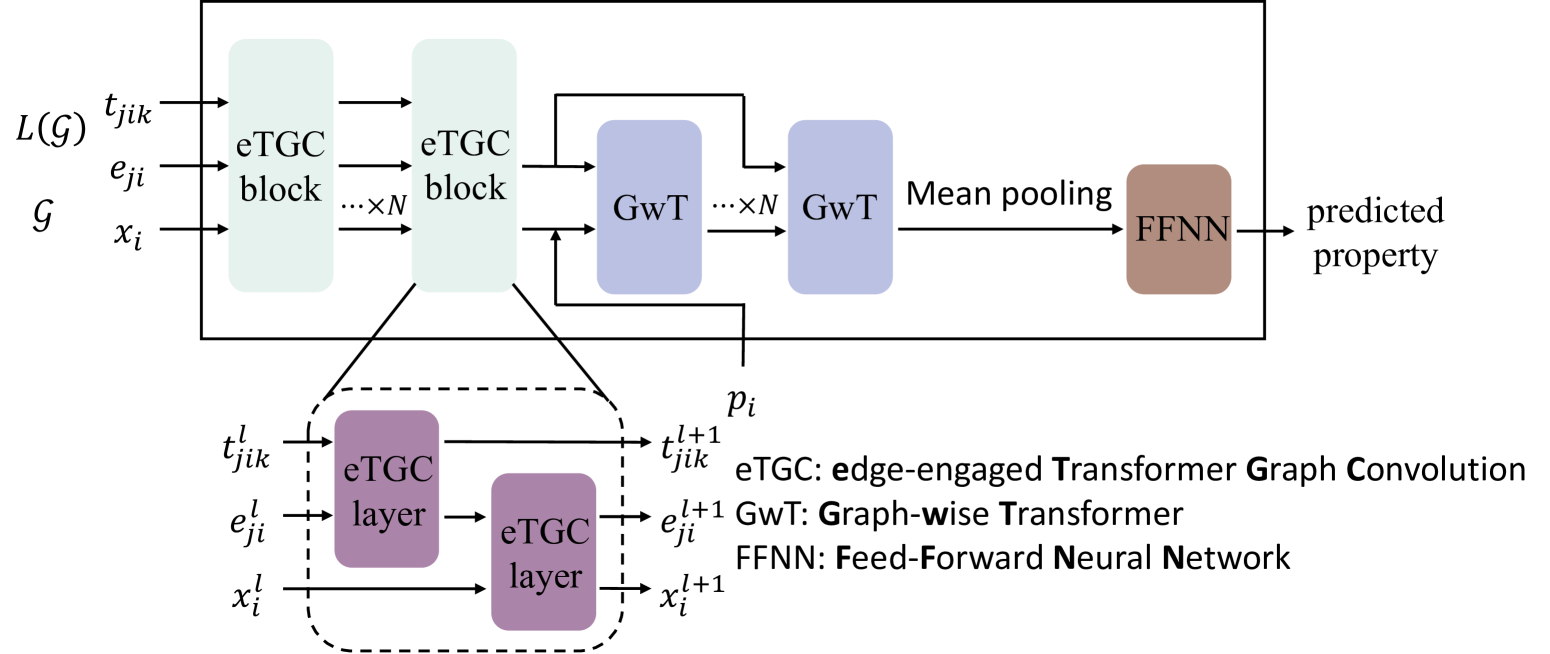
Hongyi Wang, Ji Sun, Jinzhe Liang, Li Zhai, Zitian Tang, Zijian Li, Wei Zhai, Xusheng Wang, Weihao Gao, Sheng Gong, Bolong Huang, Hua Zhang
The ionic bonding across the lattice and ordered microscopic structures endow crystals with unique symmetry and determine their macroscopic properties. Unconventional crystals, in particular, exhibit non-traditional lattice structures or possess exotic physical properties, making them intriguing subjects for investigation. Therefore, to accurately predict the physical and chemical properties of crystals, it is crucial to consider long-range orders. While GNN excels at capturing the local environment of atoms in crystals, they often face challenges in effectively capturing longer-ranged interactions due to their limited depth. In this paper, we propose CrysToGraph ($textbf{Crys}$tals with $textbf{T}$ransformers $textbf{o}$n $textbf{Graph}$s), a novel transformer-based geometric graph network designed specifically for unconventional crystalline systems, and UnconvBench, a comprehensive benchmark to evaluate models' predictive performance on unconventional crystal materials such as defected crystals, low-dimension crystals and MOF. CrysToGraph effectively captures short-range interactions with transformer-based graph convolution blocks as well as long-range interactions with graph-wise transformer blocks. CrysToGraph proofs its effectiveness in modelling unconventional crystal materials in multiple tasks, and moreover, it outperforms most existing methods, achieving new state-of-the-art results on the benchmarks of both unconventional crystals and traditional crystals.
Read more7/24/2024
0
Space Group Informed Transformer for Crystalline Materials Generation
Zhendong Cao, Xiaoshan Luo, Jian Lv, Lei Wang
We introduce CrystalFormer, a transformer-based autoregressive model specifically designed for space group-controlled generation of crystalline materials. The incorporation of space group symmetry significantly simplifies the crystal space, which is crucial for data and compute efficient generative modeling of crystalline materials. Leveraging the prominent discrete and sequential nature of the Wyckoff positions, CrystalFormer learns to generate crystals by directly predicting the species and locations of symmetry-inequivalent atoms in the unit cell. We demonstrate the advantages of CrystalFormer in standard tasks such as symmetric structure initialization and element substitution compared to conventional methods implemented in popular crystal structure prediction software. Moreover, we showcase the application of CrystalFormer of property-guided materials design in a plug-and-play manner. Our analysis shows that CrystalFormer ingests sensible solid-state chemistry knowledge and heuristics by compressing the material dataset, thus enabling systematic exploration of crystalline materials. The simplicity, generality, and flexibility of CrystalFormer position it as a promising architecture to be the foundational model of the entire crystalline materials space, heralding a new era in materials modeling and discovery.
Read more8/19/2024
🤿
0
Structure to Property: Chemical Element Embeddings and a Deep Learning Approach for Accurate Prediction of Chemical Properties
Shokirbek Shermukhamedov, Dilorom Mamurjonova, Michael Probst
We introduce the elEmBERT model for chemical classification tasks. It is based on deep learning techniques, such as a multilayer encoder architecture. We demonstrate the opportunities offered by our approach on sets of organic, inorganic and crystalline compounds. In particular, we developed and tested the model using the Matbench and Moleculenet benchmarks, which include crystal properties and drug design-related benchmarks. We also conduct an analysis of vector representations of chemical compounds, shedding light on the underlying patterns in structural data. Our model exhibits exceptional predictive capabilities and proves universally applicable to molecular and material datasets. For instance, on the Tox21 dataset, we achieved an average precision of 96%, surpassing the previously best result by 10%.
Read more8/20/2024