AsEP: Benchmarking Deep Learning Methods for Antibody-specific Epitope Prediction

0

Sign in to get full access
Overview
- This paper presents a benchmark study on deep learning methods for predicting antibody-specific epitopes (AsEP).
- The researchers evaluated the performance of several deep learning models on a dataset of antibody-antigen protein complexes.
- The goal was to identify the most effective approaches for predicting epitopes that are recognized by specific antibodies.
Plain English Explanation
Antibodies and Epitopes
Antibodies are proteins produced by the immune system that can recognize and bind to specific molecules, called antigens, on the surface of pathogens or foreign substances. The specific region on an antigen that an antibody binds to is called an epitope.
Predicting Antibody-Specific Epitopes
Accurately predicting the epitopes that a particular antibody will recognize is important for developing effective vaccines and treatments. This paper evaluates different deep learning methods for tackling this challenge.
The Benchmark Study
The researchers tested several deep learning models on a dataset of antibody-antigen complexes. They wanted to see which approaches were most effective at predicting the epitopes that would be recognized by a given antibody.
Technical Explanation
The paper evaluated the performance of various deep learning architectures, including convolutional neural networks (CNNs), transformers, and graph neural networks, for the task of antibody-specific epitope prediction. The models were trained and tested on a dataset of over 2,000 antibody-antigen protein complex structures.
Key features of the models included:
- Representing the antibody-antigen complex as a graph, with amino acid residues as nodes and interactions as edges
- Using transformer-based architectures to capture long-range dependencies in the protein structures
- Incorporating both sequence and structural information about the antibody and antigen
The researchers compared the performance of the deep learning models to traditional structure-based epitope prediction methods, demonstrating significant improvements in accuracy.
Critical Analysis
The paper provides a comprehensive benchmark of deep learning approaches for antibody-specific epitope prediction, which is an important problem in computational biology and immunology. The authors thoroughly evaluate a range of model architectures and feature representations, offering valuable insights into the most effective techniques.
However, the dataset used in the study, while sizable, may not fully capture the diversity of antibody-antigen interactions found in the real world. Additionally, the paper does not explore the potential for active learning or multi-modal approaches to further improve the predictive performance.
Conclusion
This paper presents a significant advancement in the field of antibody-specific epitope prediction, demonstrating the power of deep learning techniques for this important problem. The benchmark results provide a valuable reference for researchers and developers working on related challenges in computational immunology and protein structure prediction.
This summary was produced with help from an AI and may contain inaccuracies - check out the links to read the original source documents!
Related Papers
0
AsEP: Benchmarking Deep Learning Methods for Antibody-specific Epitope Prediction
Chunan Liu, Lilian Denzler, Yihong Chen, Andrew Martin, Brooks Paige
Epitope identification is vital for antibody design yet challenging due to the inherent variability in antibodies. While many deep learning methods have been developed for general protein binding site prediction tasks, whether they work for epitope prediction remains an understudied research question. The challenge is also heightened by the lack of a consistent evaluation pipeline with sufficient dataset size and epitope diversity. We introduce a filtered antibody-antigen complex structure dataset, AsEP (Antibody-specific Epitope Prediction). AsEP is the largest of its kind and provides clustered epitope groups, allowing the community to develop and test novel epitope prediction methods. AsEP comes with an easy-to-use interface in Python and pre-built graph representations of each antibody-antigen complex while also supporting customizable embedding methods. Based on this new dataset, we benchmarked various representative general protein-binding site prediction methods and find that their performances are not satisfactory as expected for epitope prediction. We thus propose a new method, WALLE, that leverages both protein language models and graph neural networks. WALLE demonstrate about 5X performance gain over existing methods. Our empirical findings evidence that epitope prediction benefits from combining sequential embeddings provided by language models and geometrical information from graph representations, providing a guideline for future method design. In addition, we reformulate the task as bipartite link prediction, allowing easy model performance attribution and interpretability. We open-source our data and code at https://github.com/biochunan/AsEP-dataset.
Read more7/26/2024

0
Improving Paratope and Epitope Prediction by Multi-Modal Contrastive Learning and Interaction Informativeness Estimation
Zhiwei Wang, Yongkang Wang, Wen Zhang
Accurately predicting antibody-antigen binding residues, i.e., paratopes and epitopes, is crucial in antibody design. However, existing methods solely focus on uni-modal data (either sequence or structure), disregarding the complementary information present in multi-modal data, and most methods predict paratopes and epitopes separately, overlooking their specific spatial interactions. In this paper, we propose a novel Multi-modal contrastive learning and Interaction informativeness estimation-based method for Paratope and Epitope prediction, named MIPE, by using both sequence and structure data of antibodies and antigens. MIPE implements a multi-modal contrastive learning strategy, which maximizes representations of binding and non-binding residues within each modality and meanwhile aligns uni-modal representations towards effective modal representations. To exploit the spatial interaction information, MIPE also incorporates an interaction informativeness estimation that computes the estimated interaction matrices between antibodies and antigens, thereby approximating them to the actual ones. Extensive experiments demonstrate the superiority of our method compared to baselines. Additionally, the ablation studies and visualizations demonstrate the superiority of MIPE owing to the better representations acquired through multi-modal contrastive learning and the interaction patterns comprehended by the interaction informativeness estimation.
Read more6/3/2024
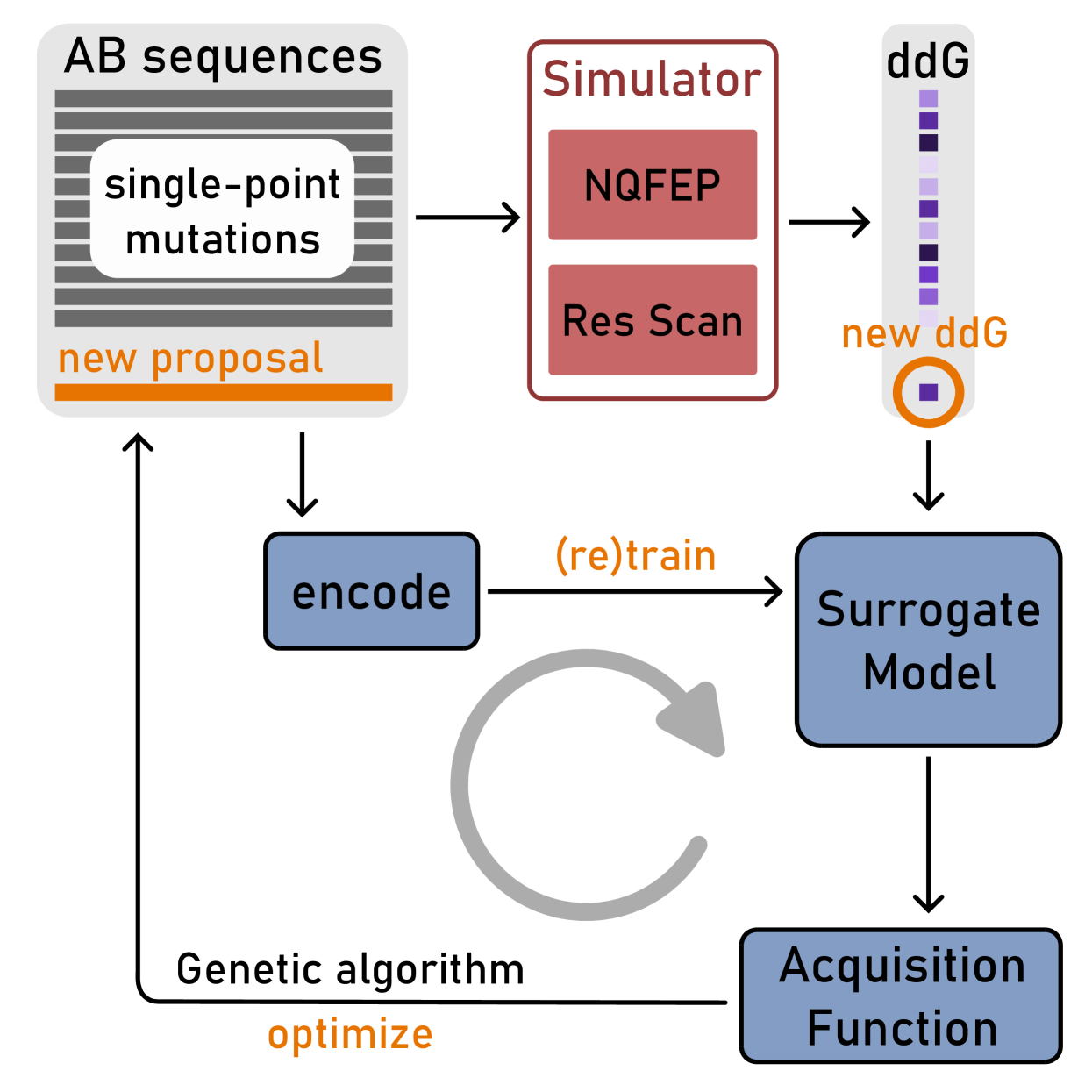
0
Active learning for affinity prediction of antibodies
Alexandra Gessner, Sebastian W. Ober, Owen Vickery, Dino Ogli'c, Talip Uc{c}ar
The primary objective of most lead optimization campaigns is to enhance the binding affinity of ligands. For large molecules such as antibodies, identifying mutations that enhance antibody affinity is particularly challenging due to the combinatorial explosion of potential mutations. When the structure of the antibody-antigen complex is available, relative binding free energy (RBFE) methods can offer valuable insights into how different mutations will impact the potency and selectivity of a drug candidate, thereby reducing the reliance on costly and time-consuming wet-lab experiments. However, accurately simulating the physics of large molecules is computationally intensive. We present an active learning framework that iteratively proposes promising sequences for simulators to evaluate, thereby accelerating the search for improved binders. We explore different modeling approaches to identify the most effective surrogate model for this task, and evaluate our framework both using pre-computed pools of data and in a realistic full-loop setting.
Read more6/12/2024

0
Topology-enhanced machine learning model (Top-ML) for anticancer peptide prediction
Joshua Zhi En Tan, JunJie Wee, Xue Gong, Kelin Xia
Recently, therapeutic peptides have demonstrated great promise for cancer treatment. To explore powerful anticancer peptides, artificial intelligence (AI)-based approaches have been developed to systematically screen potential candidates. However, the lack of efficient featurization of peptides has become a bottleneck for these machine-learning models. In this paper, we propose a topology-enhanced machine learning model (Top-ML) for anticancer peptide prediction. Our Top-ML employs peptide topological features derived from its sequence connection information characterized by vector and spectral descriptors. Our Top-ML model has been validated on two widely used AntiCP 2.0 benchmark datasets and has achieved state-of-the-art performance. Our results highlight the potential of leveraging novel topology-based featurization to accelerate the identification of anticancer peptides.
Read more7/15/2024