A deep graph model for the signed interaction prediction in biological network

0

Sign in to get full access
Overview
- This paper presents a deep graph model for predicting signed interactions in biological networks.
- The model leverages graph neural networks to capture the complex relationships between nodes (e.g., proteins, genes) and their interactions, which can be either positive or negative.
- The proposed approach aims to improve the accuracy of predicting whether two nodes in a biological network will have a positive or negative interaction.
Plain English Explanation
Biological systems like the human body are incredibly complex, with various components (e.g., proteins, genes) interacting in intricate ways. These interactions can be either positive (e.g., two proteins working together) or negative (e.g., one protein inhibiting another). Accurately predicting the nature of these interactions is crucial for understanding how biological systems function and developing treatments for diseases.
The researchers in this paper developed a deep learning model that can analyze the structure of biological networks and predict whether the interactions between different components will be positive or negative. The model uses a specialized type of neural network called a graph neural network, which is particularly well-suited for capturing the complex, interconnected relationships in biological data.
By leveraging this advanced deep learning approach, the researchers were able to improve the accuracy of predicting the sign (positive or negative) of interactions in biological networks compared to previous methods. This could have important applications in fields like drug discovery and network medicine, where understanding the complex relationships between biological components is key to developing effective treatments.
Technical Explanation
The researchers developed a deep graph model for the signed interaction prediction problem in biological networks. Their approach, called SigGNN, uses a graph neural network (GNN) architecture to capture the complex relationships between nodes (e.g., proteins, genes) and their interactions, which can be either positive or negative.
The key components of the SigGNN model include:
-
Graph Encoder: This module takes the input biological network and encodes the nodes and edges into a higher-dimensional representation using multiple GNN layers.
-
Interaction Predictor: This module takes the encoded node representations and predicts the sign (positive or negative) of the interactions between pairs of nodes.
-
Contrastive Learning: The researchers also incorporated a contrastive learning approach to enhance the model's ability to learn meaningful representations of the node interactions.
The researchers evaluated their SigGNN model on several real-world biological network datasets and compared its performance to state-of-the-art baselines. Their results showed that SigGNN outperformed the other methods in accurately predicting the sign of interactions, demonstrating the effectiveness of the deep graph modeling approach for this task.
Critical Analysis
The researchers acknowledge several limitations and areas for future work in their paper. For example, they note that their model primarily focuses on the network structure and does not incorporate additional biological features, such as node attributes or domain knowledge. Incorporating such information could potentially further improve the model's performance.
Additionally, the researchers suggest that extending the SigGNN model to handle dynamic changes in biological networks over time could be a valuable direction for future research. This could be particularly important for understanding how biological systems evolve and respond to various stimuli.
While the researchers have demonstrated the effectiveness of their deep graph modeling approach, it would be beneficial to see further validation of the model's performance on larger and more diverse biological network datasets. This could help establish the generalizability of the approach and its potential impact on real-world applications.
Conclusion
This paper presents a novel deep graph model, SigGNN, for predicting the sign (positive or negative) of interactions in biological networks. By leveraging graph neural networks, the model is able to capture the complex relationships between the components of biological systems more effectively than previous methods.
The improved accuracy of the SigGNN model in predicting the nature of interactions could have important implications for fields like drug discovery and network medicine, where understanding the intricate web of biological interactions is crucial for developing effective treatments. The researchers have also highlighted several promising directions for future work, such as incorporating additional biological features and exploring dynamic network analysis.
Overall, this research demonstrates the power of advanced deep learning techniques, like graph neural networks, in advancing our understanding of complex biological systems and paving the way for more effective interventions and therapies.
This summary was produced with help from an AI and may contain inaccuracies - check out the links to read the original source documents!
Related Papers
0
A deep graph model for the signed interaction prediction in biological network
Shuyi Jin, Mengji Zhang, Meijie Wang, Lun Yu
In pharmaceutical research, the strategy of drug repurposing accelerates the development of new therapies while reducing R&D costs. Network pharmacology lays the theoretical groundwork for identifying new drug indications, and deep graph models have become essential for their precision in mapping complex biological networks. Our study introduces an advanced graph model that utilizes graph convolutional networks and tensor decomposition to effectively predict signed chemical-gene interactions. This model demonstrates superior predictive performance, especially in handling the polar relations in biological networks. Our research opens new avenues for drug discovery and repurposing, especially in understanding the mechanism of actions of drugs.
Read more7/11/2024
🧠
0
DDoS: A Graph Neural Network based Drug Synergy Prediction Algorithm
Kyriakos Schwarz, Alicia Pliego-Mendieta, Amina Mollaysa, Lara Planas-Paz, Chantal Pauli, Ahmed Allam, Michael Krauthammer
Drug synergy arises when the combined impact of two drugs exceeds the sum of their individual effects. While single-drug effects on cell lines are well-documented, the scarcity of data on drug synergy, considering the vast array of potential drug combinations, prompts a growing interest in computational approaches for predicting synergies in untested drug pairs. We introduce a Graph Neural Network (textit{GNN}) based model for drug synergy prediction, which utilizes drug chemical structures and cell line gene expression data. We extract data from the largest available drug combination database (DrugComb) and generate multiple synergy scores (commonly used in the literature) to create seven datasets that serve as a reliable benchmark with high confidence. In contrast to conventional models relying on pre-computed chemical features, our GNN-based approach learns task-specific drug representations directly from the graph structure of the drugs, providing superior performance in predicting drug synergies. Our work suggests that learning task-specific drug representations and leveraging a diverse dataset is a promising approach to advancing our understanding of drug-drug interaction and synergy.
Read more4/29/2024
🔮
0
Research on Adverse Drug Reaction Prediction Model Combining Knowledge Graph Embedding and Deep Learning
Yufeng Li, Wenchao Zhao, Bo Dang, Xu Yan, Weimin Wang, Min Gao, Mingxuan Xiao
In clinical treatment, identifying potential adverse reactions of drugs can help assist doctors in making medication decisions. In response to the problems in previous studies that features are high-dimensional and sparse, independent prediction models need to be constructed for each adverse reaction of drugs, and the prediction accuracy is low, this paper develops an adverse drug reaction prediction model based on knowledge graph embedding and deep learning, which can predict experimental results. Unified prediction of adverse drug reactions covered. Knowledge graph embedding technology can fuse the associated information between drugs and alleviate the shortcomings of high-dimensional sparsity in feature matrices, and the efficient training capabilities of deep learning can improve the prediction accuracy of the model. This article builds an adverse drug reaction knowledge graph based on drug feature data; by analyzing the embedding effect of the knowledge graph under different embedding strategies, the best embedding strategy is selected to obtain sample vectors; and then a convolutional neural network model is constructed to predict adverse reactions. The results show that under the DistMult embedding model and 400-dimensional embedding strategy, the convolutional neural network model has the best prediction effect; the average accuracy, F_1 score, recall rate and area under the curve of repeated experiments are better than the methods reported in the literature. The obtained prediction model has good prediction accuracy and stability, and can provide an effective reference for later safe medication guidance.
Read more7/30/2024
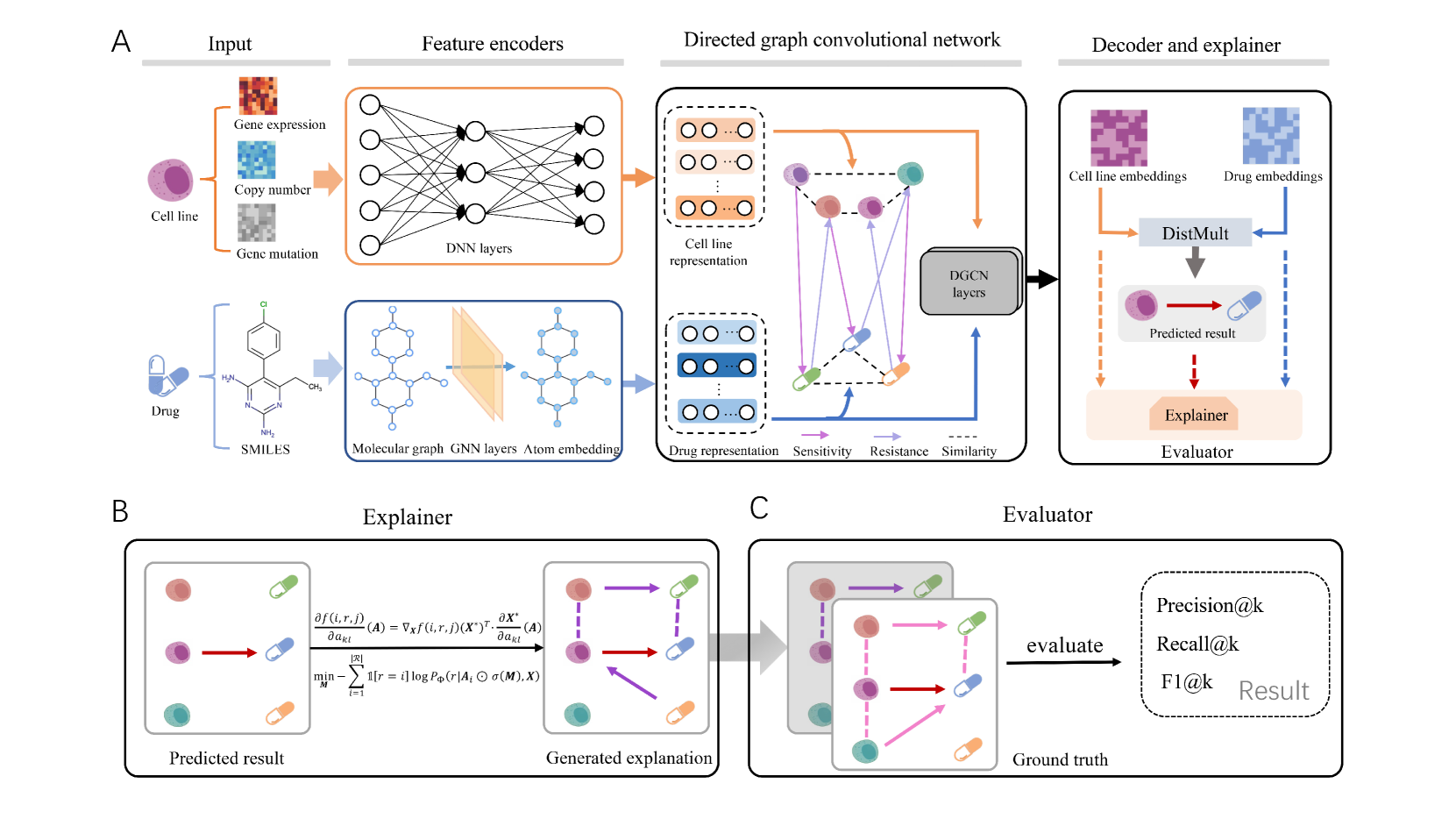
0
DRExplainer: Quantifiable Interpretability in Drug Response Prediction with Directed Graph Convolutional Network
Haoyuan Shi, Tao Xu, Xiaodi Li, Qian Gao, Junfeng Xia, Zhenyu Yue
Predicting the response of a cancer cell line to a therapeutic drug is pivotal for personalized medicine. Despite numerous deep learning methods that have been developed for drug response prediction, integrating diverse information about biological entities and predicting the directional response remain major challenges. Here, we propose a novel interpretable predictive model, DRExplainer, which leverages a directed graph convolutional network to enhance the prediction in a directed bipartite network framework. DRExplainer constructs a directed bipartite network integrating multi-omics profiles of cell lines, the chemical structure of drugs and known drug response to achieve directed prediction. Then, DRExplainer identifies the most relevant subgraph to each prediction in this directed bipartite network by learning a mask, facilitating critical medical decision-making. Additionally, we introduce a quantifiable method for model interpretability that leverages a ground truth benchmark dataset curated from biological features. In computational experiments, DRExplainer outperforms state-of-the-art predictive methods and another graph-based explanation method under the same experimental setting. Finally, the case studies further validate the interpretability and the effectiveness of DRExplainer in predictive novel drug response. Our code is available at: https://github.com/vshy-dream/DRExplainer.
Read more8/23/2024