Diff4VS: HIV-inhibiting Molecules Generation with Classifier Guidance Diffusion for Virtual Screening

0
Sign in to get full access
Overview
- This paper introduces Diff4VS, a framework that uses a diffusion model guided by a classifier to generate HIV-inhibiting molecules for virtual screening.
- The key contributions are:
- Incorporating a classifier-guided diffusion model to generate HIV-inhibiting molecules.
- Leveraging generated molecules for virtual screening to identify potential drug candidates.
- Demonstrating the effectiveness of the approach on multiple HIV target proteins.
Plain English Explanation
The paper presents a new approach to help find potential drug molecules that can inhibit the HIV virus. The researchers developed a machine learning model called Diff4VS that can
The key idea is to use a type of machine learning model called a "diffusion model" that can learn to create new molecules from scratch. However, the researchers added an extra component - a "classifier" model that can identify whether a molecule is likely to be good at inhibiting HIV. This classifier model is used to
The researchers tested this approach on several different HIV target proteins, and showed that the generated molecules could be useful for
Technical Explanation
The Diff4VS framework consists of two main components:
-
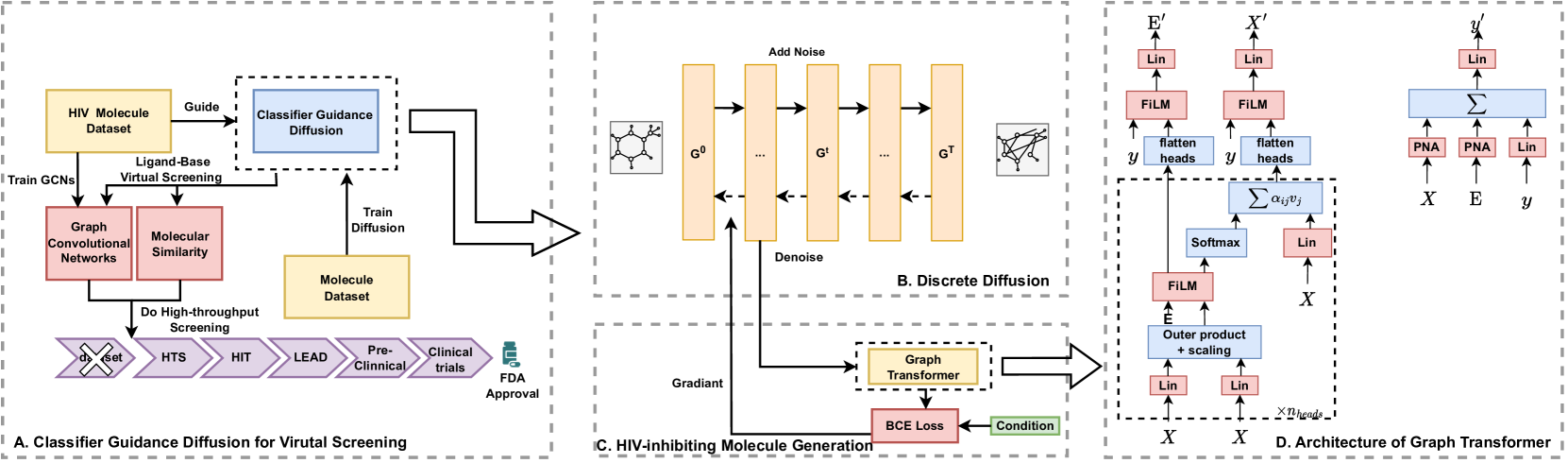
Classifier-Guided Diffusion Model: The core of the system is a diffusion model, which is a type of generative machine learning model that can learn to create new molecules from scratch. This diffusion model is guided by a classifier model that has been trained to identify HIV-inhibiting molecules. The classifier provides feedback to the diffusion model during training, helping it generate molecules that are more likely to be effective against HIV.
-
Virtual Screening Pipeline: The generated molecules from the diffusion model are then used as input to a virtual screening pipeline. Virtual screening computationally evaluates the potential of these molecules to bind to and inhibit target HIV proteins, identifying the most promising candidates for further exploration and experimental validation.
The researchers evaluated the Diff4VS framework on multiple HIV target proteins, demonstrating its ability to generate diverse sets of HIV-inhibiting molecules that could be useful starting points for drug discovery. The generated molecules were shown to have favorable drug-like properties and exhibited strong binding affinities to the target proteins in the virtual screening experiments.
Critical Analysis
The paper provides a compelling approach for leveraging generative AI models to accelerate the drug discovery process for HIV. The key strength is the integration of a classifier-guided diffusion model, which allows the system to focus on generating molecules that are more likely to be effective against the target proteins.
However, the paper does not extensively discuss the limitations of the approach. For example, it's unclear how the generated molecules would perform in real-world experimental validation, and what challenges might arise in transitioning from virtual screening to actual drug development. Additionally, the paper does not explore the potential for the framework to be applied to other disease targets beyond HIV.
Further research is needed to better understand the broader applicability and scalability of the Diff4VS approach, as well as its long-term impact on accelerating the drug discovery process.
Conclusion
The Diff4VS framework represents a promising advance in the use of generative AI models for computer-aided drug design. By incorporating a classifier-guided diffusion model, the system can generate diverse sets of HIV-inhibiting molecules that show potential for virtual screening and further drug development. While more research is needed to fully evaluate the approach, this work highlights the exciting potential of combining cutting-edge machine learning techniques with domain-specific knowledge to streamline the complex and time-consuming drug discovery process.
This summary was produced with help from an AI and may contain inaccuracies - check out the links to read the original source documents!
Related Papers
0
Diff4VS: HIV-inhibiting Molecules Generation with Classifier Guidance Diffusion for Virtual Screening
Jiaqing Lyu, Changjie Chen, Bing Liang, Yijia Zhang
The AIDS epidemic has killed 40 million people and caused serious global problems. The identification of new HIV-inhibiting molecules is of great importance for combating the AIDS epidemic. Here, the Classifier Guidance Diffusion model and ligand-based virtual screening strategy are combined to discover potential HIV-inhibiting molecules for the first time. We call it Diff4VS. An extra classifier is trained using the HIV molecule dataset, and the gradient of the classifier is used to guide the Diffusion to generate HIV-inhibiting molecules. Experiments show that Diff4VS can generate more candidate HIV-inhibiting molecules than other methods. Inspired by ligand-based virtual screening, a new metric DrugIndex is proposed. The DrugIndex is the ratio of the proportion of candidate drug molecules in the generated molecule to the proportion of candidate drug molecules in the training set. DrugIndex provides a new evaluation method for evolving molecular generative models from a pharmaceutical perspective. Besides, we report a new phenomenon observed when using molecule generation models for virtual screening. Compared to real molecules, the generated molecules have a lower proportion that is highly similar to known drug molecules. We call it Degradation in molecule generation. Based on the data analysis, the Degradation may result from the difficulty of generating molecules with a specific structure in the generative model. Our research contributes to the application of generative models in drug design from method, metric, and phenomenon analysis.
Read more7/24/2024
➖
0
DiffBP: Generative Diffusion of 3D Molecules for Target Protein Binding
Haitao Lin, Yufei Huang, Odin Zhang, Siqi Ma, Meng Liu, Xuanjing Li, Lirong Wu, Jishui Wang, Tingjun Hou, Stan Z. Li
Generating molecules that bind to specific proteins is an important but challenging task in drug discovery. Previous works usually generate atoms in an auto-regressive way, where element types and 3D coordinates of atoms are generated one by one. However, in real-world molecular systems, the interactions among atoms in an entire molecule are global, leading to the energy function pair-coupled among atoms. With such energy-based consideration, the modeling of probability should be based on joint distributions, rather than sequentially conditional ones. Thus, the unnatural sequentially auto-regressive modeling of molecule generation is likely to violate the physical rules, thus resulting in poor properties of the generated molecules. In this work, a generative diffusion model for molecular 3D structures based on target proteins as contextual constraints is established, at a full-atom level in a non-autoregressive way. Given a designated 3D protein binding site, our model learns the generative process that denoises both element types and 3D coordinates of an entire molecule, with an equivariant network. Experimentally, the proposed method shows competitive performance compared with prevailing works in terms of high affinity with proteins and appropriate molecule sizes as well as other drug properties such as drug-likeness of the generated molecules.
Read more7/16/2024

0
Synthetic Data from Diffusion Models Improve Drug Discovery Prediction
Bing Hu, Ashish Saragadam, Anita Layton, Helen Chen
Artificial intelligence (AI) is increasingly used in every stage of drug development. Continuing breakthroughs in AI-based methods for drug discovery require the creation, improvement, and refinement of drug discovery data. We posit a new data challenge that slows the advancement of drug discovery AI: datasets are often collected independently from each other, often with little overlap, creating data sparsity. Data sparsity makes data curation difficult for researchers looking to answer key research questions requiring values posed across multiple datasets. We propose a novel diffusion GNN model Syngand capable of generating ligand and pharmacokinetic data end-to-end. We show and provide a methodology for sampling pharmacokinetic data for existing ligands using our Syngand model. We show the initial promising results on the efficacy of the Syngand-generated synthetic target property data on downstream regression tasks with AqSolDB, LD50, and hERG central. Using our proposed model and methodology, researchers can easily generate synthetic ligand data to help them explore research questions that require data spanning multiple datasets.
Read more5/8/2024

0
Molecular Diffusion Models with Virtual Receptors
Matan Halfon, Eyal Rozenberg, Ehud Rivlin, Daniel Freedman
Machine learning approaches to Structure-Based Drug Design (SBDD) have proven quite fertile over the last few years. In particular, diffusion-based approaches to SBDD have shown great promise. We present a technique which expands on this diffusion approach in two crucial ways. First, we address the size disparity between the drug molecule and the target/receptor, which makes learning more challenging and inference slower. We do so through the notion of a Virtual Receptor, which is a compressed version of the receptor; it is learned so as to preserve key aspects of the structural information of the original receptor, while respecting the relevant group equivariance. Second, we incorporate a protein language embedding used originally in the context of protein folding. We experimentally demonstrate the contributions of both the virtual receptors and the protein embeddings: in practice, they lead to both better performance, as well as significantly faster computations.
Read more6/27/2024