Generative Active Learning for the Search of Small-molecule Protein Binders

0

Sign in to get full access
Overview
- This research paper explores a novel approach called "Generative Active Learning" (GAL) to efficiently search for small-molecule protein binders.
- The key idea is to train a generative model to explore the vast chemical space and identify promising drug candidates, guided by active learning techniques that provide feedback to the model.
- The researchers demonstrate the effectiveness of this approach on various protein targets, showing that GAL can outperform traditional methods in discovering high-affinity binders.
Plain English Explanation
Developing new drugs is a complex and challenging process. Researchers often start by searching through millions of potential drug-like molecules to find ones that can effectively bind to and interact with a target protein. This paper introduces a new approach called "Generative Active Learning" (GAL) that aims to streamline this search process.
The core idea is to train a machine learning model that can
The researchers demonstrate that this GAL approach outperforms traditional methods on several real-world protein targets. By combining powerful generative models with active learning, GAL can navigate the complex landscape of potential drug molecules more effectively than manual search or random sampling.
Technical Explanation
The researchers propose a Generative Active Learning (GAL) framework for the search of small-molecule protein binders. The key components are:
- A generative model, specifically a Variational Autoencoder (VAE), that can generate new drug-like molecules by learning the underlying distribution of chemical space.
- An active learning loop, where the generative model proposes candidate molecules, which are then evaluated for binding affinity to the target protein. The feedback from these evaluations is used to fine-tune the generative model, guiding it to focus on more promising regions of chemical space.
The researchers evaluated GAL on several protein targets and compared its performance to traditional methods like random sampling and Bayesian optimization. The results show that GAL can consistently identify high-affinity binders more efficiently, requiring fewer experimental evaluations to find good candidates.
The success of GAL stems from its ability to effectively explore the vast chemical space by leveraging the generative model, while also incorporating feedback to continuously refine the search. This allows GAL to navigate the complex landscape of potential drug molecules in a more targeted and efficient manner compared to traditional approaches.
Critical Analysis
The researchers acknowledge several limitations and areas for future work. First, the performance of GAL is dependent on the quality of the generative model, which may struggle to accurately capture the underlying distribution of drug-like molecules. Improvements to the generative model architecture or training techniques could further enhance the effectiveness of the GAL approach.
Additionally, the researchers only evaluated GAL on a limited set of protein targets. Further testing on a broader range of targets, including more challenging cases, would help validate the general applicability of the method.
Another potential issue is the reliance on experimental binding affinity evaluations, which can be time-consuming and costly. Integrating computational methods for faster initial screening could help reduce the experimental burden and make GAL more scalable.
Overall, the Generative Active Learning approach presented in this paper represents a promising direction for accelerating the search for small-molecule protein binders. By combining generative modeling and active learning, it offers a more efficient alternative to traditional drug discovery methods.
Conclusion
This research introduces a novel Generative Active Learning (GAL) framework for the search of small-molecule protein binders. The key innovation is the use of a generative model to explore the vast chemical space, guided by active learning techniques that provide feedback to continuously refine the search.
The results demonstrate that GAL can outperform traditional methods in identifying high-affinity binders for various protein targets, requiring fewer experimental evaluations. This highlights the potential of combining powerful generative models with active learning to navigate the complex landscape of potential drug molecules more effectively.
While the approach has some limitations that require further research, the Generative Active Learning framework represents an exciting step forward in accelerating the drug discovery process and expanding the repertoire of available small-molecule therapeutics.
This summary was produced with help from an AI and may contain inaccuracies - check out the links to read the original source documents!
Related Papers
0
Generative Active Learning for the Search of Small-molecule Protein Binders
Maksym Korablyov, Cheng-Hao Liu, Moksh Jain, Almer M. van der Sloot, Eric Jolicoeur, Edward Ruediger, Andrei Cristian Nica, Emmanuel Bengio, Kostiantyn Lapchevskyi, Daniel St-Cyr, Doris Alexandra Schuetz, Victor Ion Butoi, Jarrid Rector-Brooks, Simon Blackburn, Leo Feng, Hadi Nekoei, SaiKrishna Gottipati, Priyesh Vijayan, Prateek Gupta, Ladislav Ramp'av{s}ek, Sasikanth Avancha, Pierre-Luc Bacon, William L. Hamilton, Brooks Paige, Sanchit Misra, Stanislaw Kamil Jastrzebski, Bharat Kaul, Doina Precup, Jos'e Miguel Hern'andez-Lobato, Marwin Segler, Michael Bronstein, Anne Marinier, Mike Tyers, Yoshua Bengio
Despite substantial progress in machine learning for scientific discovery in recent years, truly de novo design of small molecules which exhibit a property of interest remains a significant challenge. We introduce LambdaZero, a generative active learning approach to search for synthesizable molecules. Powered by deep reinforcement learning, LambdaZero learns to search over the vast space of molecules to discover candidates with a desired property. We apply LambdaZero with molecular docking to design novel small molecules that inhibit the enzyme soluble Epoxide Hydrolase 2 (sEH), while enforcing constraints on synthesizability and drug-likeliness. LambdaZero provides an exponential speedup in terms of the number of calls to the expensive molecular docking oracle, and LambdaZero de novo designed molecules reach docking scores that would otherwise require the virtual screening of a hundred billion molecules. Importantly, LambdaZero discovers novel scaffolds of synthesizable, drug-like inhibitors for sEH. In in vitro experimental validation, a series of ligands from a generated quinazoline-based scaffold were synthesized, and the lead inhibitor N-(4,6-di(pyrrolidin-1-yl)quinazolin-2-yl)-N-methylbenzamide (UM0152893) displayed sub-micromolar enzyme inhibition of sEH.
Read more5/6/2024
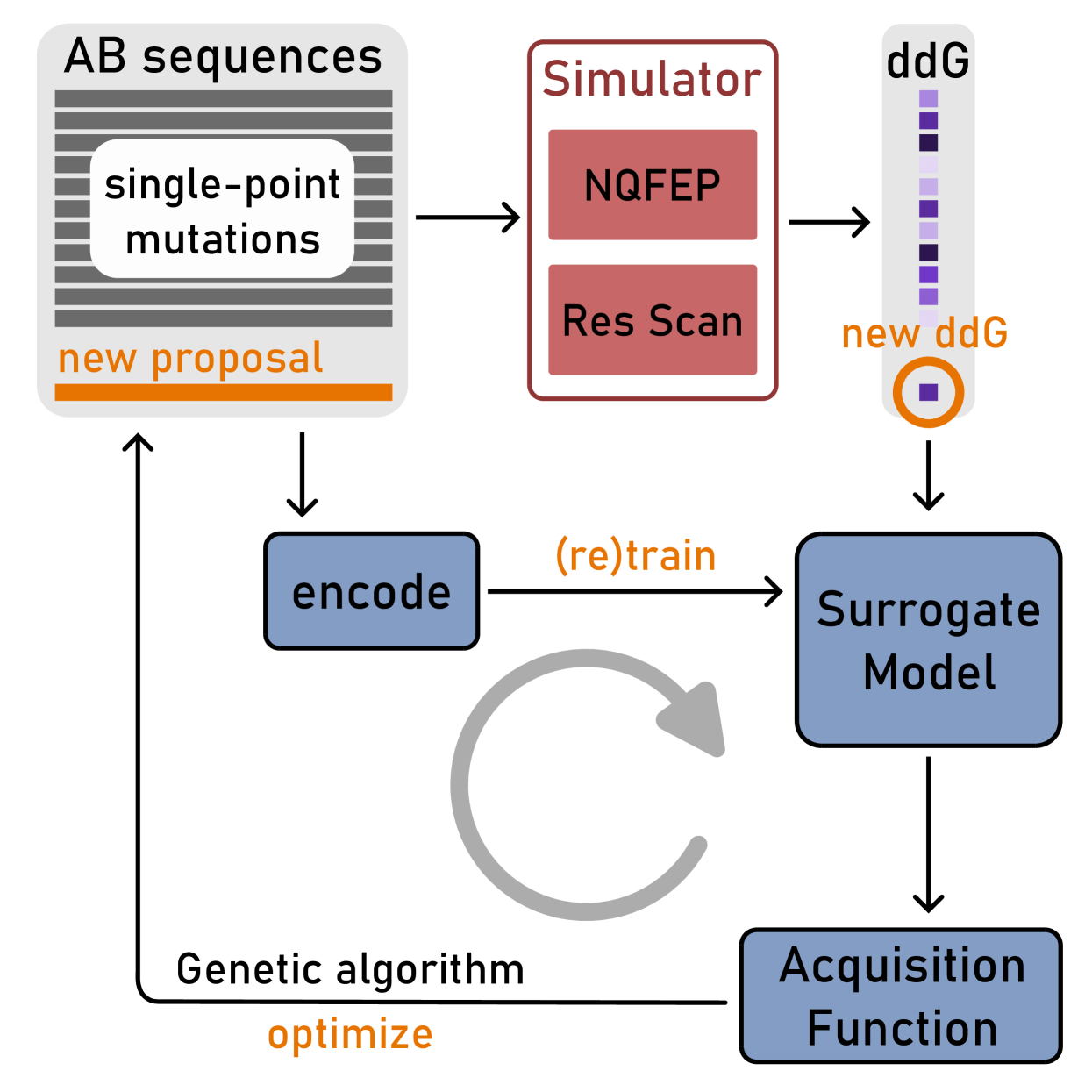
0
Active learning for affinity prediction of antibodies
Alexandra Gessner, Sebastian W. Ober, Owen Vickery, Dino Ogli'c, Talip Uc{c}ar
The primary objective of most lead optimization campaigns is to enhance the binding affinity of ligands. For large molecules such as antibodies, identifying mutations that enhance antibody affinity is particularly challenging due to the combinatorial explosion of potential mutations. When the structure of the antibody-antigen complex is available, relative binding free energy (RBFE) methods can offer valuable insights into how different mutations will impact the potency and selectivity of a drug candidate, thereby reducing the reliance on costly and time-consuming wet-lab experiments. However, accurately simulating the physics of large molecules is computationally intensive. We present an active learning framework that iteratively proposes promising sequences for simulators to evaluate, thereby accelerating the search for improved binders. We explore different modeling approaches to identify the most effective surrogate model for this task, and evaluate our framework both using pre-computed pools of data and in a realistic full-loop setting.
Read more6/12/2024

0
Generative Model for Small Molecules with Latent Space RL Fine-Tuning to Protein Targets
Ulrich A. Mbou Sob, Qiulin Li, Miguel Arbes'u, Oliver Bent, Andries P. Smit, Arnu Pretorius
A specific challenge with deep learning approaches for molecule generation is generating both syntactically valid and chemically plausible molecular string representations. To address this, we propose a novel generative latent-variable transformer model for small molecules that leverages a recently proposed molecular string representation called SAFE. We introduce a modification to SAFE to reduce the number of invalid fragmented molecules generated during training and use this to train our model. Our experiments show that our model can generate novel molecules with a validity rate > 90% and a fragmentation rate < 1% by sampling from a latent space. By fine-tuning the model using reinforcement learning to improve molecular docking, we significantly increase the number of hit candidates for five specific protein targets compared to the pre-trained model, nearly doubling this number for certain targets. Additionally, our top 5% mean docking scores are comparable to the current state-of-the-art (SOTA), and we marginally outperform SOTA on three of the five targets.
Read more7/22/2024

0
Generative Enzyme Design Guided by Functionally Important Sites and Small-Molecule Substrates
Zhenqiao Song, Yunlong Zhao, Wenxian Shi, Wengong Jin, Yang Yang, Lei Li
Enzymes are genetically encoded biocatalysts capable of accelerating chemical reactions. How can we automatically design functional enzymes? In this paper, we propose EnzyGen, an approach to learn a unified model to design enzymes across all functional families. Our key idea is to generate an enzyme's amino acid sequence and their three-dimensional (3D) coordinates based on functionally important sites and substrates corresponding to a desired catalytic function. These sites are automatically mined from enzyme databases. EnzyGen consists of a novel interleaving network of attention and neighborhood equivariant layers, which captures both long-range correlation in an entire protein sequence and local influence from nearest amino acids in 3D space. To learn the generative model, we devise a joint training objective, including a sequence generation loss, a position prediction loss and an enzyme-substrate interaction loss. We further construct EnzyBench, a dataset with 3157 enzyme families, covering all available enzymes within the protein data bank (PDB). Experimental results show that our EnzyGen consistently achieves the best performance across all 323 testing families, surpassing the best baseline by 10.79% in terms of substrate binding affinity. These findings demonstrate EnzyGen's superior capability in designing well-folded and effective enzymes binding to specific substrates with high affinities.
Read more7/18/2024