Heterogeneous Causal Metapath Graph Neural Network for Gene-Microbe-Disease Association Prediction

0
Sign in to get full access
Related Work
Heterogeneous Network and Graph Attention Auto-Encoder for lncRNA-Disease Association Prediction
Several recent studies have explored the use of heterogeneous networks and graph attention mechanisms for predicting associations between long non-coding RNAs (lncRNAs) and diseases. These approaches leverage the complex relationships between different biological entities, such as genes, proteins, and diseases, to improve the accuracy of predictions.
LncRNA-Disease Association Prediction Method Based on Heterogeneous Network
Another related work focuses on developing a heterogeneous network-based method for predicting lncRNA-disease associations. This approach integrates multiple data sources, including lncRNA-gene, gene-disease, and lncRNA-disease associations, to construct a comprehensive network and apply graph-based algorithms for inference.
Pathology Genomic Fusion via Biologically Informed Cross-Modal Attention
Researchers have also explored the use of cross-modal attention mechanisms to fuse genomic and pathological data for disease prediction tasks. This approach leverages the complementary information from different data modalities to enhance the model's performance.
Applying BioBeRT to Extract Germline Gene-Disease Associations
The use of pre-trained language models, such as BioBeRT, has been investigated for the task of extracting germline gene-disease associations from biomedical literature. This technique can help automate the process of identifying relevant associations from a large corpus of scientific publications.
Investigation of Customized Medical Decision Algorithms Utilizing Graph
Graph-based approaches have also been applied to the development of customized medical decision algorithms. These techniques leverage the rich relational information in healthcare data to provide personalized recommendations and insights for clinical decision-making.
Plain English Explanation
The research paper focuses on developing a novel graph neural network model, called Heterogeneous Causal Metapath Graph Neural Network (HCMGNN), for predicting associations between genes, microbes, and diseases. This task is important for understanding the complex interactions within the human body and how they contribute to various health conditions.
The key idea behind HCMGNN is to explicitly model the causal relationships between different biological entities, using a graph-based representation. The model leverages heterogeneous data sources, such as gene-disease, microbe-disease, and gene-microbe associations, to construct a comprehensive network. It then applies a causal metapath-based approach to capture the directional influence between these entities.
By incorporating the causal structure of the relationships, the model can better infer the underlying mechanisms that drive the connections between genes, microbes, and diseases. This is a significant improvement over previous approaches that relied on more generic network representations or did not explicitly consider the causal nature of the associations.
The researchers demonstrate the effectiveness of HCMGNN through extensive experiments on real-world datasets, showing improved performance in predicting gene-microbe-disease associations compared to state-of-the-art methods. This advance has the potential to contribute to a deeper understanding of the complex interplay between these biological entities and aid in the development of more targeted and effective interventions for various health conditions.
Technical Explanation
The paper proposes a Heterogeneous Causal Metapath Graph Neural Network (HCMGNN) model for predicting gene-microbe-disease associations. The key aspects of the model are as follows:
-
Heterogeneous Network Construction: The researchers integrate multiple data sources, including gene-disease, microbe-disease, and gene-microbe associations, to construct a comprehensive heterogeneous network. This network captures the complex relationships between different biological entities.
-
Causal Metapath Extraction: The model leverages causal metapaths to capture the directional influence between genes, microbes, and diseases. Metapaths are sequences of node and edge types that represent specific types of relationships in the heterogeneous network.
-
Causal Metapath-based Graph Attention: The model employs a causal metapath-based graph attention mechanism to learn the importance of different metapaths when aggregating information from the neighborhood of each node. This allows the model to focus on the most relevant causal relationships.
-
Causal Metapath-based Embedding: The learned node embeddings from the causal metapath-based graph attention are then used to predict the gene-microbe-disease associations.
The researchers evaluate the performance of HCMGNN on real-world datasets and compare it to several state-of-the-art methods. The results demonstrate the effectiveness of the proposed approach in predicting gene-microbe-disease associations, outperforming baseline models that do not explicitly consider the causal nature of the relationships.
Critical Analysis
The paper presents a well-designed and comprehensive approach for predicting gene-microbe-disease associations using a heterogeneous causal metapath-based graph neural network. The key strength of the proposed HCMGNN model is its ability to explicitly capture the directional influence between different biological entities, which is a crucial aspect of understanding the underlying mechanisms driving these associations.
However, the paper could have addressed several limitations and potential areas for further research. For example, the authors do not discuss the robustness of the model to noisy or incomplete data, which is a common challenge in real-world biological datasets. Additionally, the paper could have explored the interpretability of the learned causal metapaths and how they align with existing biological knowledge, which could provide valuable insights for domain experts.
Furthermore, the researchers could have investigated the transferability of the HCMGNN model to other related tasks, such as predicting drug-target interactions or identifying novel therapeutic targets, to demonstrate the broader applicability of the approach.
Despite these potential areas for improvement, the paper presents a significant contribution to the field of computational biology, demonstrating the power of integrating causal reasoning with graph neural networks for uncovering complex biological associations.
Conclusion
The Heterogeneous Causal Metapath Graph Neural Network (HCMGNN) proposed in this research paper represents a notable advancement in the field of gene-microbe-disease association prediction. By explicitly modeling the causal relationships between these biological entities, the model can better capture the underlying mechanisms driving their interactions, leading to improved predictive performance.
The integration of heterogeneous data sources and the novel causal metapath-based graph attention mechanism are the key innovations that set HCMGNN apart from previous approaches. These advancements have the potential to provide valuable insights into the complex interplay between genes, microbes, and diseases, which could aid in the development of more targeted and effective interventions for various health conditions.
While the paper highlights several strengths of the proposed model, it also identifies potential avenues for further research, such as exploring the robustness to noisy data and the transferability of the approach to other related tasks. Addressing these limitations could further strengthen the HCMGNN model and its applicability in the broader field of computational biology.
Overall, this research paper represents a significant step forward in understanding the intricate relationships between genes, microbes, and diseases, and the authors' work has the potential to significantly impact the field and contribute to advancements in personalized medicine and targeted therapies.
This summary was produced with help from an AI and may contain inaccuracies - check out the links to read the original source documents!
Related Papers
0
Heterogeneous Causal Metapath Graph Neural Network for Gene-Microbe-Disease Association Prediction
Kexin Zhang, Feng Huang, Luotao Liu, Zhankun Xiong, Hongyu Zhang, Yuan Quan, Wen Zhang
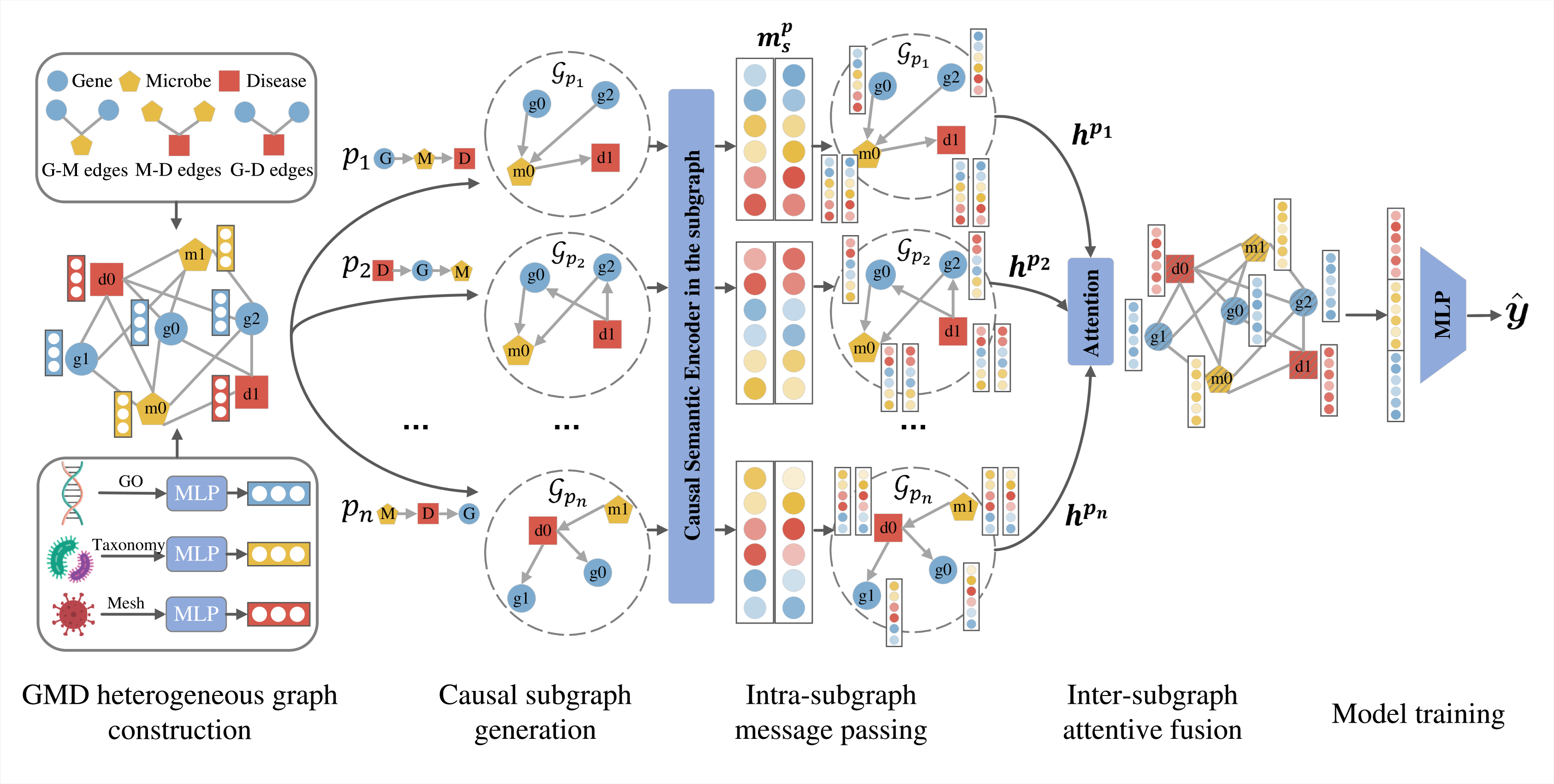
The recent focus on microbes in human medicine highlights their potential role in the genetic framework of diseases. To decode the complex interactions among genes, microbes, and diseases, computational predictions of gene-microbe-disease (GMD) associations are crucial. Existing methods primarily address gene-disease and microbe-disease associations, but the more intricate triple-wise GMD associations remain less explored. In this paper, we propose a Heterogeneous Causal Metapath Graph Neural Network (HCMGNN) to predict GMD associations. HCMGNN constructs a heterogeneous graph linking genes, microbes, and diseases through their pairwise associations, and utilizes six predefined causal metapaths to extract directed causal subgraphs, which facilitate the multi-view analysis of causal relations among three entity types. Within each subgraph, we employ a causal semantic sharing message passing network for node representation learning, coupled with an attentive fusion method to integrate these representations for predicting GMD associations. Our extensive experiments show that HCMGNN effectively predicts GMD associations and addresses association sparsity issue by enhancing the graph's semantics and structure.
Read more6/28/2024

0
Graph Neural Networks for Gut Microbiome Metaomic data: A preliminary work
Christopher Irwin, Flavio Mignone, Stefania Montani, Luigi Portinale
The gut microbiome, crucial for human health, presents challenges in analyzing its complex metaomic data due to high dimensionality and sparsity. Traditional methods struggle to capture its intricate relationships. We investigate graph neural networks (GNNs) for this task, aiming to derive meaningful representations of individual gut microbiomes. Unlike methods relying solely on taxa abundance, we directly leverage phylogenetic relationships, in order to obtain a generalized encoder for taxa networks. The representation learnt from the encoder are then used to train a model for phenotype prediction such as Inflammatory Bowel Disease (IBD).
Read more7/2/2024
🌐
0
Heterogeneous network and graph attention auto-encoder for LncRNA-disease association prediction
Jin-Xing Liu, Wen-Yu Xi, Ling-Yun Dai, Chun-Hou Zheng, Ying-Lian Gao
The emerging research shows that lncRNAs are associated with a series of complex human diseases. However, most of the existing methods have limitations in identifying nonlinear lncRNA-disease associations (LDAs), and it remains a huge challenge to predict new LDAs. Therefore, the accurate identification of LDAs is very important for the warning and treatment of diseases. In this work, multiple sources of biomedical data are fully utilized to construct characteristics of lncRNAs and diseases, and linear and nonlinear characteristics are effectively integrated. Furthermore, a novel deep learning model based on graph attention automatic encoder is proposed, called HGATELDA. To begin with, the linear characteristics of lncRNAs and diseases are created by the miRNA-lncRNA interaction matrix and miRNA-disease interaction matrix. Following this, the nonlinear features of diseases and lncRNAs are extracted using a graph attention auto-encoder, which largely retains the critical information and effectively aggregates the neighborhood information of nodes. In the end, LDAs can be predicted by fusing the linear and nonlinear characteristics of diseases and lncRNA. The HGATELDA model achieves an impressive AUC value of 0.9692 when evaluated using a 5-fold cross-validation indicating its superior performance in comparison to several recent prediction models. Meanwhile, the effectiveness of HGATELDA in identifying novel LDAs is further demonstrated by case studies. the HGATELDA model appears to be a viable computational model for predicting LDAs.
Read more5/7/2024
🔮
0
LncRNA-disease association prediction method based on heterogeneous information completion and convolutional neural network
Wen-Yu Xi, Juan Wang, Yu-Lin Zhang, Jin-Xing Liu, Yin-Lian Gao
The emerging research shows that lncRNA has crucial research value in a series of complex human diseases. Therefore, the accurate identification of lncRNA-disease associations (LDAs) is very important for the warning and treatment of diseases. However, most of the existing methods have limitations in identifying nonlinear LDAs, and it remains a huge challenge to predict new LDAs. In this paper, a deep learning model based on a heterogeneous network and convolutional neural network (CNN) is proposed for lncRNA-disease association prediction, named HCNNLDA. The heterogeneous network containing the lncRNA, disease, and miRNA nodes, is constructed firstly. The embedding matrix of a lncRNA-disease node pair is constructed according to various biological premises about lncRNAs, diseases, and miRNAs. Then, the low-dimensional feature representation is fully learned by the convolutional neural network. In the end, the XGBoot classifier model is trained to predict the potential LDAs. HCNNLDA obtains a high AUC value of 0.9752 and AUPR of 0.9740 under the 5-fold cross-validation. The experimental results show that the proposed model has better performance than that of several latest prediction models. Meanwhile, the effectiveness of HCNNLDA in identifying novel LDAs is further demonstrated by case studies of three diseases. To sum up, HCNNLDA is a feasible calculation model to predict LDAs.
Read more6/6/2024