Quantum-inspired Reinforcement Learning for Synthesizable Drug Design

0
🏅
Sign in to get full access
Overview
- The research paper discusses a novel approach to drug design using quantum-inspired reinforcement learning.
- The proposed method aims to generate synthesizable drug candidates with desirable properties.
- The research leverages quantum computing principles and reinforcement learning techniques to optimize the drug design process.
Plain English Explanation
The researchers have developed a new way to design potential new drugs using a combination of quantum computing ideas and a machine learning technique called reinforcement learning. The goal is to create drug molecules that not only have the right properties to be effective medicines, but are also easy to manufacture or "synthesize" in a lab.
Traditionally, drug discovery has been a slow and expensive process, involving a lot of trial and error. The researchers believe that by using quantum computing principles and reinforcement learning, they can make this process more efficient and targeted. The key idea is to train a reinforcement learning algorithm to explore the vast space of possible drug molecules, and guide it towards ones that are both effective and synthesizable.
The paper describes the technical details of how they implemented this approach. Importantly, they show that it can generate promising drug candidates that outperform those created using more conventional methods. This suggests that quantum-inspired reinforcement learning could be a powerful tool for accelerating drug discovery in the future.
Technical Explanation
The paper proposes a quantum-inspired reinforcement learning approach for synthesizable drug design. The core idea is to leverage principles from quantum computing, such as superposition and entanglement, to enhance the exploration capabilities of a reinforcement learning agent.
The authors first define a molecular representation that encodes both the chemical structure and key physicochemical properties of drug candidates. They then train a deep reinforcement learning agent to navigate this representation, guided by a reward function that encourages the discovery of molecules with high predicted activity and synthesizability.
The quantum-inspired aspect comes from incorporating a variational quantum circuit into the agent's policy network. This allows the agent to learn a more expressive policy that can better exploit the structure of the molecular search space.
The researchers evaluate their approach on several drug discovery benchmarks, demonstrating that it outperforms conventional reinforcement learning methods in generating high-quality, synthesizable drug candidates. They also provide detailed analysis of the learned policies and discuss the potential advantages of the quantum-inspired techniques.
Critical Analysis
The paper presents a compelling approach to addressing the key challenges in drug discovery - namely, the need to find molecules that are both effective and feasible to manufacture. By incorporating quantum computing principles, the authors aim to enhance the exploration capabilities of the reinforcement learning agent, allowing it to more efficiently navigate the vast space of possible drug candidates.
However, the authors acknowledge several limitations of their work. First, the actual quantum hardware required to fully realize the quantum-inspired aspects may not be readily available, limiting the practical deployment of this method. Additionally, the paper focuses on relatively simple molecular benchmarks, and it remains to be seen how the approach would scale to more complex, real-world drug discovery problems.
Further research is also needed to better understand the specific mechanisms by which the quantum-inspired techniques improve the performance of the reinforcement learning agent. A more detailed analysis of the learned policies and their relationship to the quantum components could provide valuable insights.
Despite these caveats, the paper represents an important step towards integrating quantum computing and reinforcement learning for advancing the field of drug discovery. As quantum hardware continues to develop, approaches like the one presented here may become increasingly viable and impactful.
Conclusion
This research paper introduces a novel quantum-inspired reinforcement learning method for synthesizable drug design. By leveraging principles from quantum computing, the authors have developed a reinforcement learning agent that can more effectively explore the vast space of possible drug molecules, identifying candidates that are both pharmacologically active and feasible to manufacture.
The results demonstrate the potential of this approach to accelerate the drug discovery process, addressing a critical challenge in the field. As quantum computing technology continues to advance, techniques like the one presented in this paper may become increasingly valuable tools for researchers and drug companies working to develop new and improved medicines.
This summary was produced with help from an AI and may contain inaccuracies - check out the links to read the original source documents!
Related Papers
🏅
0
Quantum-inspired Reinforcement Learning for Synthesizable Drug Design
Dannong Wang, Jintai Chen, Zhiding Liang, Tianfan Fu, Xiao-Yang Liu
Synthesizable molecular design (also known as synthesizable molecular optimization) is a fundamental problem in drug discovery, and involves designing novel molecular structures to improve their properties according to drug-relevant oracle functions (i.e., objective) while ensuring synthetic feasibility. However, existing methods are mostly based on random search. To address this issue, in this paper, we introduce a novel approach using the reinforcement learning method with quantum-inspired simulated annealing policy neural network to navigate the vast discrete space of chemical structures intelligently. Specifically, we employ a deterministic REINFORCE algorithm using policy neural networks to output transitional probability to guide state transitions and local search using genetic algorithm to refine solutions to a local optimum within each iteration. Our methods are evaluated with the Practical Molecular Optimization (PMO) benchmark framework with a 10K query budget. We further showcase the competitive performance of our method by comparing it against the state-of-the-art genetic algorithms-based method.
Read more9/17/2024

0
Improving Targeted Molecule Generation through Language Model Fine-Tuning Via Reinforcement Learning
Salma J. Ahmed, Mustafa A. Elattar
Developing new drugs is laborious and costly, demanding extensive time investment. In this study, we introduce an innovative de-novo drug design strategy, which harnesses the capabilities of language models to devise targeted drugs for specific proteins. Employing a Reinforcement Learning (RL) framework utilizing Proximal Policy Optimization (PPO), we refine the model to acquire a policy for generating drugs tailored to protein targets. Our method integrates a composite reward function, combining considerations of drug-target interaction and molecular validity. Following RL fine-tuning, our approach demonstrates promising outcomes, yielding notable improvements in molecular validity, interaction efficacy, and critical chemical properties, achieving 65.37 for Quantitative Estimation of Drug-likeness (QED), 321.55 for Molecular Weight (MW), and 4.47 for Octanol-Water Partition Coefficient (logP), respectively. Furthermore, out of the generated drugs, only 0.041% do not exhibit novelty.
Read more5/14/2024
0
New!Hybrid quantum cycle generative adversarial network for small molecule generation
Matvei Anoshin, Asel Sagingalieva, Christopher Mansell, Dmitry Zhiganov, Vishal Shete, Markus Pflitsch, Alexey Melnikov
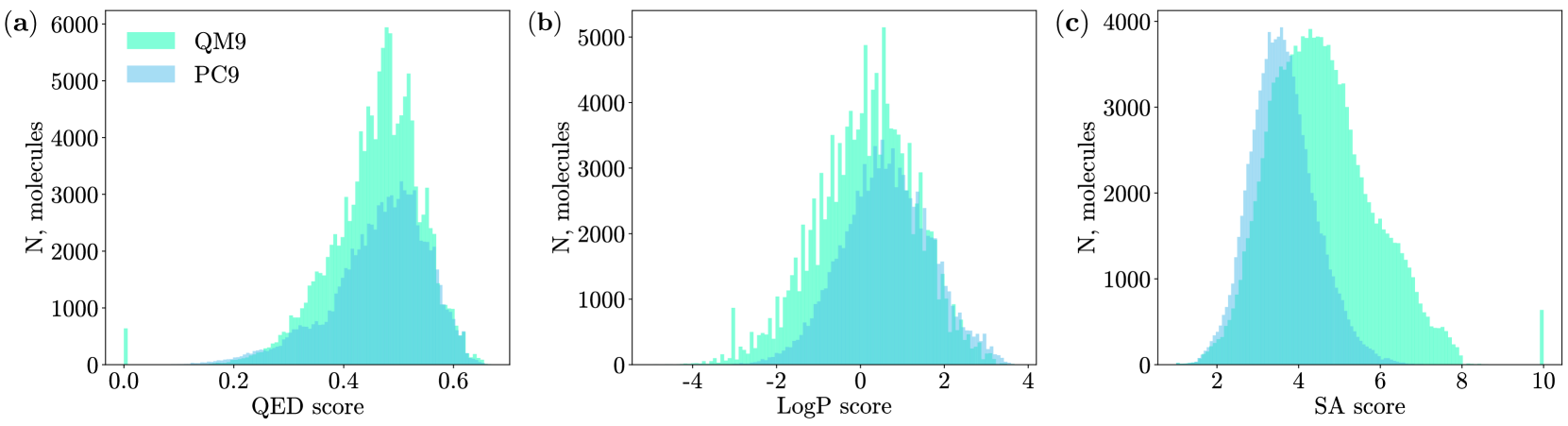
The drug design process currently requires considerable time and resources to develop each new compound that enters the market. This work develops an application of hybrid quantum generative models based on the integration of parametrized quantum circuits into known molecular generative adversarial networks, and proposes quantum cycle architectures that improve model performance and stability during training. Through extensive experimentation on benchmark drug design datasets, QM9 and PC9, the introduced models are shown to outperform the previously achieved scores. Most prominently, the new scores indicate an increase of up to 30% in the quantitative estimation of druglikeness. The new hybrid quantum machine learning algorithms, as well as the achieved scores of pharmacokinetic properties, contribute to the development of fast and accurate drug discovery processes.
Read more9/20/2024
👀
0
Implementation of The Future of Drug Discovery: QuantumBased Machine Learning Simulation (QMLS)
Yifan Zhou, Yan Shing Liang, Yew Kee Wong, Haichuan Qiu, Yu Xi Wu, Bin He
The Research & Development (R&D) phase of drug development is a lengthy and costly process. To revolutionize this process, we introduce our new concept QMLS to shorten the whole R&D phase to three to six months and decrease the cost to merely fifty to eighty thousand USD. For Hit Generation, Machine Learning Molecule Generation (MLMG) generates possible hits according to the molecular structure of the target protein while the Quantum Simulation (QS) filters molecules from the primary essay based on the reaction and binding effectiveness with the target protein. Then, For Lead Optimization, the resultant molecules generated and filtered from MLMG and QS are compared, and molecules that appear as a result of both processes will be made into dozens of molecular variations through Machine Learning Molecule Variation (MLMV), while others will only be made into a few variations. Lastly, all optimized molecules would undergo multiple rounds of QS filtering with a high standard for reaction effectiveness and safety, creating a few dozen pre-clinical-trail-ready drugs. This paper is based on our first paper, where we pitched the concept of machine learning combined with quantum simulations. In this paper we will go over the detailed design and framework of QMLS, including MLMG, MLMV, and QS.
Read more9/6/2024