RGFN: Synthesizable Molecular Generation Using GFlowNets
2406.08506
0
0
Abstract
Generative models hold great promise for small molecule discovery, significantly increasing the size of search space compared to traditional in silico screening libraries. However, most existing machine learning methods for small molecule generation suffer from poor synthesizability of candidate compounds, making experimental validation difficult. In this paper we propose Reaction-GFlowNet (RGFN), an extension of the GFlowNet framework that operates directly in the space of chemical reactions, thereby allowing out-of-the-box synthesizability while maintaining comparable quality of generated candidates. We demonstrate that with the proposed set of reactions and building blocks, it is possible to obtain a search space of molecules orders of magnitude larger than existing screening libraries coupled with low cost of synthesis. We also show that the approach scales to very large fragment libraries, further increasing the number of potential molecules. We demonstrate the effectiveness of the proposed approach across a range of oracle models, including pretrained proxy models and GPU-accelerated docking.
Create account to get full access
Overview
- This paper introduces RGFN, a method for generating synthesizable molecular compounds using a type of deep learning model called a GFlowNet.
- GFlowNets are a novel approach to generative modeling that can learn to produce complex structures, like molecules, in a step-by-step fashion.
- The RGFN method aims to generate molecules that not only have desirable properties, but also have feasible synthesis pathways.
Plain English Explanation
The paper describes a new way to use deep learning to design new chemical compounds, or molecules. The key idea is to use a special type of deep learning model called a GFlowNet. GFlowNets can learn to build complex structures, like molecules, one step at a time.
Typically, when designing new molecules, researchers have to consider both the desired properties of the molecule (e.g. it binds well to a target protein) and whether it can actually be synthesized in a lab. The RGFN method tries to address both of these challenges at once. It learns to generate molecules that not only have the right properties, but also have a clear step-by-step synthesis pathway that would allow a chemist to actually make the molecule.
This is an important advance, as it can be very difficult to design molecules that meet all the necessary criteria. The RGFN method provides a way to streamline the molecule design process and increase the chances of generating compounds that are both effective and synthesizable.
Technical Explanation
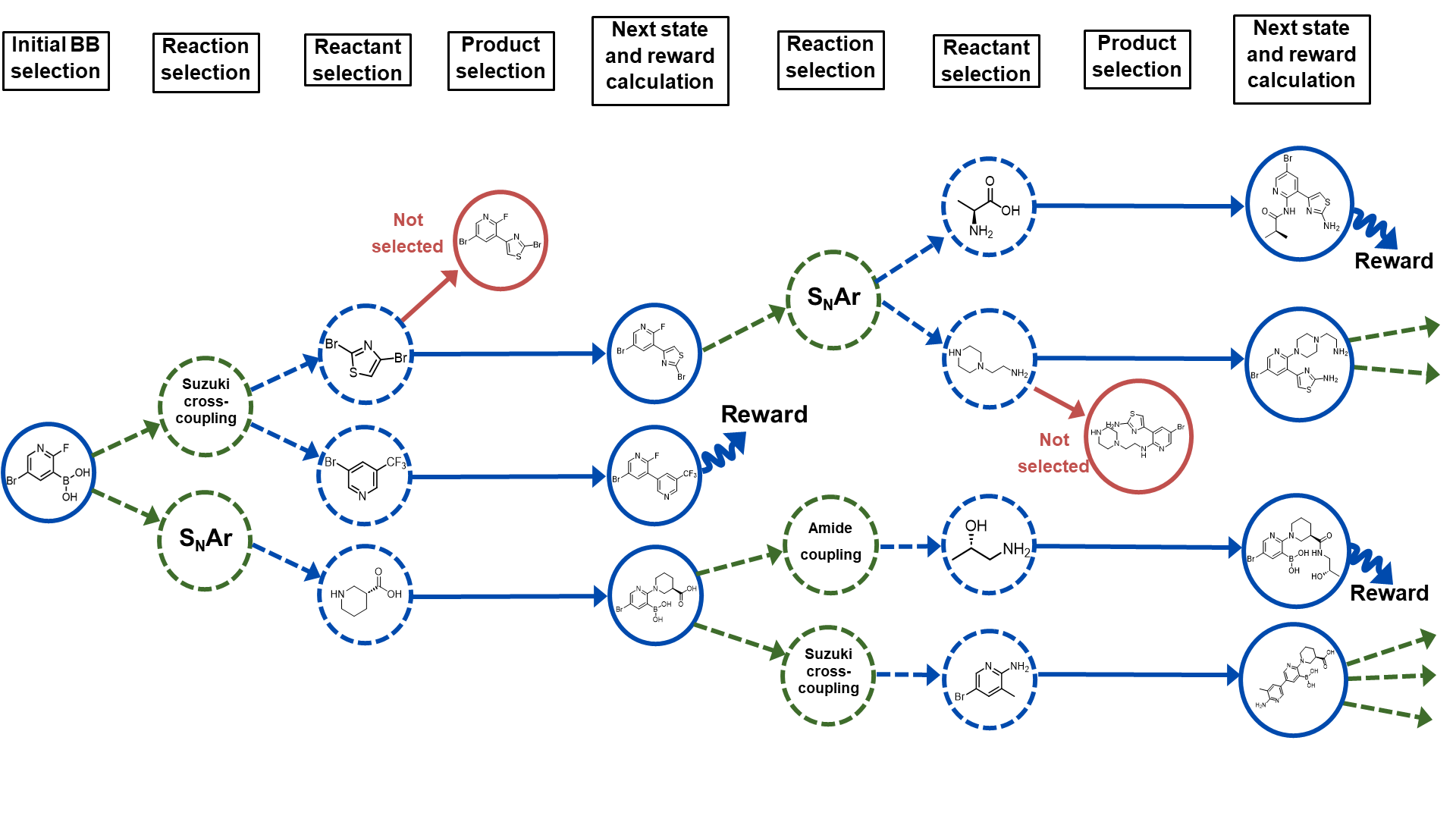
The core of the RGFN method is a GFlowNet, a type of generative model that learns to build complex structures in a step-by-step fashion. Unlike traditional generative models that produce a complete output all at once, GFlowNets learn to make a sequence of decisions that gradually assemble the final structure.
The RGFN GFlowNet is trained on a dataset of known, synthesizable molecules. It learns to mimic the process of how these molecules are built, one atom or bond at a time. Once trained, the GFlowNet can then be used to generate new molecule candidates, ensuring that each step in the synthesis process is feasible.
To guide the GFlowNet towards generating desirable molecules, the authors incorporate reinforcement learning techniques. The model receives rewards for producing molecules with certain target properties, incentivizing it to explore regions of the chemical space that are both synthesizable and have the desired characteristics.
The authors demonstrate the effectiveness of RGFN through experiments on several molecular optimization tasks. They show that RGFN can generate molecules that outperform those produced by other state-of-the-art generative models, while also ensuring the molecules have viable synthesis pathways.
Critical Analysis
The RGFN method represents an important step forward in the field of computational molecular design. By explicitly considering synthetic feasibility during the generation process, it addresses a key challenge that has limited the real-world impact of many previous generative models.
However, the paper does note some limitations of the RGFN approach. For example, the model is currently limited to generating relatively small molecules, as the complexity of the synthesis pathways increases exponentially with molecular size. Additionally, the reliance on a dataset of known, synthesizable molecules means the method may struggle to explore truly novel chemical space.
Further research will be needed to address these limitations and expand the capabilities of RGFN-style models. Potential areas for improvement include developing more efficient GFlowNet architectures, incorporating additional chemical domain knowledge, and exploring methods to learn synthesis pathways from broader data sources.
Overall, the RGFN paper makes a valuable contribution by demonstrating the potential of GFlowNets for generating synthesizable molecules. As the field of computational molecular design continues to advance, approaches like RGFN will play an important role in bridging the gap between in silico and in vitro discovery.
Conclusion
The RGFN method presented in this paper represents a significant advancement in the field of computational molecular design. By incorporating the concept of synthetic feasibility into the generative modeling process, RGFN can produce molecules that are not only desirable, but also realistically synthesizable in the laboratory.
This is an important step forward, as it helps address a key challenge that has limited the practical impact of many previous generative models for drug discovery and materials science. The RGFN approach, built upon the powerful GFlowNet framework, shows how deep learning can be leveraged to streamline the molecule design process and increase the chances of generating compounds with real-world potential.
While the current RGFN model has some limitations, the underlying ideas and techniques demonstrated in this paper lay the groundwork for continued progress in this area. As the field of computational molecular design continues to evolve, methods like RGFN will play a vital role in bridging the gap between in silico and in vitro discovery, accelerating the development of new molecules with transformative applications.
This summary was produced with help from an AI and may contain inaccuracies - check out the links to read the original source documents!
Related Papers

SynFlowNet: Towards Molecule Design with Guaranteed Synthesis Pathways
Miruna Cretu, Charles Harris, Julien Roy, Emmanuel Bengio, Pietro Li`o
0
0
Recent breakthroughs in generative modelling have led to a number of works proposing molecular generation models for drug discovery. While these models perform well at capturing drug-like motifs, they are known to often produce synthetically inaccessible molecules. This is because they are trained to compose atoms or fragments in a way that approximates the training distribution, but they are not explicitly aware of the synthesis constraints that come with making molecules in the lab. To address this issue, we introduce SynFlowNet, a GFlowNet model whose action space uses chemically validated reactions and reactants to sequentially build new molecules. We evaluate our approach using synthetic accessibility scores and an independent retrosynthesis tool. SynFlowNet consistently samples synthetically feasible molecules, while still being able to find diverse and high-utility candidates. Furthermore, we compare molecules designed with SynFlowNet to experimentally validated actives, and find that they show comparable properties of interest, such as molecular weight, SA score and predicted protein binding affinity.
5/3/2024

RetroGFN: Diverse and Feasible Retrosynthesis using GFlowNets
Piotr Gai'nski, Micha{l} Koziarski, Krzysztof Maziarz, Marwin Segler, Jacek Tabor, Marek 'Smieja
0
0
Single-step retrosynthesis aims to predict a set of reactions that lead to the creation of a target molecule, which is a crucial task in molecular discovery. Although a target molecule can often be synthesized with multiple different reactions, it is not clear how to verify the feasibility of a reaction, because the available datasets cover only a tiny fraction of the possible solutions. Consequently, the existing models are not encouraged to explore the space of possible reactions sufficiently. In this paper, we propose a novel single-step retrosynthesis model, RetroGFN, that can explore outside the limited dataset and return a diverse set of feasible reactions by leveraging a feasibility proxy model during the training. We show that RetroGFN achieves competitive results on standard top-k accuracy while outperforming existing methods on round-trip accuracy. Moreover, we provide empirical arguments in favor of using round-trip accuracy which expands the notion of feasibility with respect to the standard top-k accuracy metric.
6/28/2024
Genetic-guided GFlowNets for Sample Efficient Molecular Optimization
Hyeonah Kim, Minsu Kim, Sanghyeok Choi, Jinkyoo Park
0
0
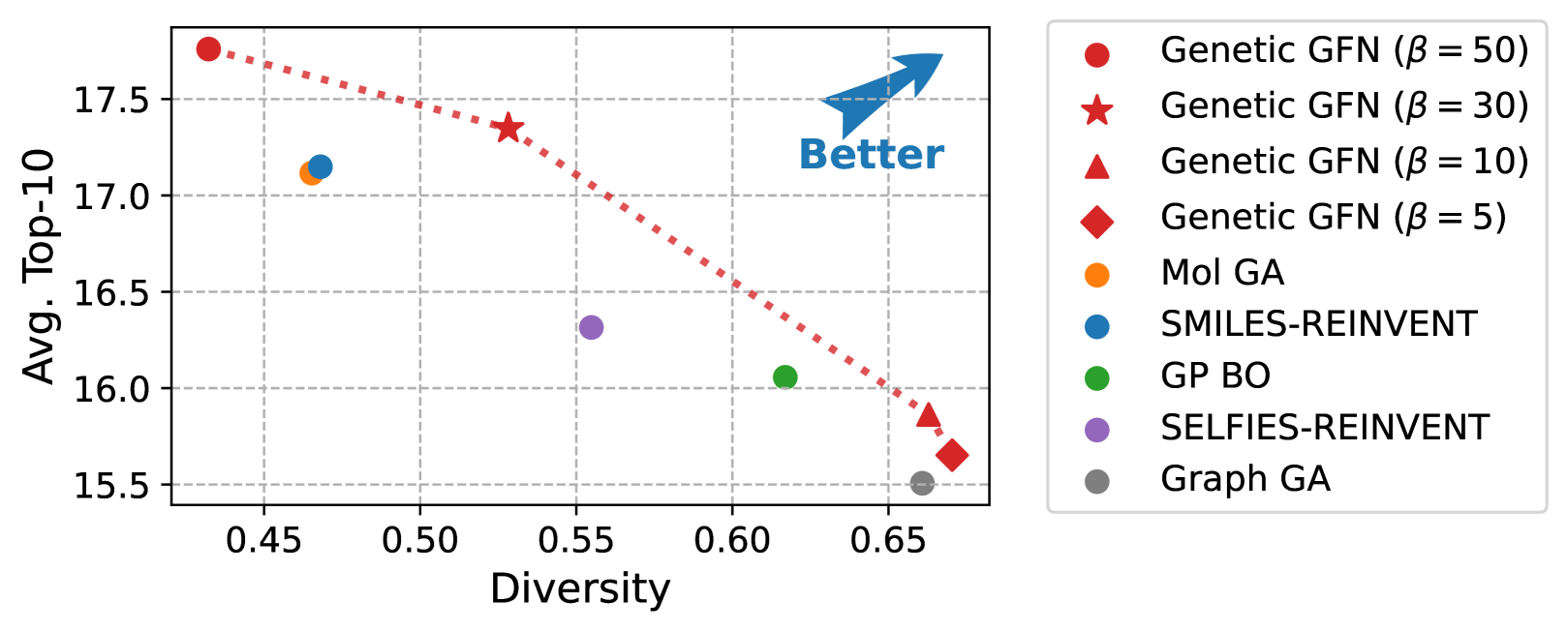
The challenge of discovering new molecules with desired properties is crucial in domains like drug discovery and material design. Recent advances in deep learning-based generative methods have shown promise but face the issue of sample efficiency due to the computational expense of evaluating the reward function. This paper proposes a novel algorithm for sample-efficient molecular optimization by distilling a powerful genetic algorithm into deep generative policy using GFlowNets training, the off-policy method for amortized inference. This approach enables the deep generative policy to learn from domain knowledge, which has been explicitly integrated into the genetic algorithm. Our method achieves state-of-the-art performance in the official molecular optimization benchmark, significantly outperforming previous methods. It also demonstrates effectiveness in designing inhibitors against SARS-CoV-2 with substantially fewer reward calls.
5/28/2024
🚀
TacoGFN: Target-conditioned GFlowNet for Structure-based Drug Design
Tony Shen, Seonghwan Seo, Grayson Lee, Mohit Pandey, Jason R Smith, Artem Cherkasov, Woo Youn Kim, Martin Ester
0
0
Searching the vast chemical space for drug-like and synthesizable molecules with high binding affinity to a protein pocket is a challenging task in drug discovery. Recently, molecular deep generative models have been introduced which promise to be more efficient than exhaustive virtual screening, by directly generating molecules based on the protein structure. However, since they learn the distribution of a limited protein-ligand complex dataset, the existing methods struggle with generating novel molecules with significant property improvements. In this paper, we frame the generation task as a Reinforcement Learning task, where the goal is to search the wider chemical space for molecules with desirable properties as opposed to fitting a training data distribution. More specifically, we propose TacoGFN, a Generative Flow Network conditioned on protein pocket structure, using binding affinity, drug-likeliness and synthesizability measures as our reward. Empirically, our method outperforms state-of-art methods on the CrossDocked2020 benchmark for every molecular property (Vina score, QED, SA), while significantly improving the generation time. TacoGFN achieves $-8.82$ in median docking score and $52.63%$ in Novel Hit Rate.
4/9/2024