TacoGFN: Target-conditioned GFlowNet for Structure-based Drug Design
2310.03223
0
0
🚀
Abstract
Searching the vast chemical space for drug-like and synthesizable molecules with high binding affinity to a protein pocket is a challenging task in drug discovery. Recently, molecular deep generative models have been introduced which promise to be more efficient than exhaustive virtual screening, by directly generating molecules based on the protein structure. However, since they learn the distribution of a limited protein-ligand complex dataset, the existing methods struggle with generating novel molecules with significant property improvements. In this paper, we frame the generation task as a Reinforcement Learning task, where the goal is to search the wider chemical space for molecules with desirable properties as opposed to fitting a training data distribution. More specifically, we propose TacoGFN, a Generative Flow Network conditioned on protein pocket structure, using binding affinity, drug-likeliness and synthesizability measures as our reward. Empirically, our method outperforms state-of-art methods on the CrossDocked2020 benchmark for every molecular property (Vina score, QED, SA), while significantly improving the generation time. TacoGFN achieves $-8.82$ in median docking score and $52.63%$ in Novel Hit Rate.
Create account to get full access
Overview
- Developing new drugs is a complex challenge, as researchers must search through vast chemical spaces to find molecules that effectively bind to target proteins.
- Traditional virtual screening methods are limited in their ability to explore this chemical space efficiently.
- Recent advances in molecular deep generative models offer a promising alternative, but these models struggle to generate truly novel molecules with significant property improvements.
- This paper proposes a new approach called TacoGFN that frames the molecule generation task as a reinforcement learning problem, aiming to explore the wider chemical space for optimal properties.
Plain English Explanation
Discovering new drugs is a bit like trying to find a needle in a haystack. Researchers have to search through an incredibly vast number of possible chemical compounds, looking for ones that can effectively bind to and interact with specific proteins in the body. This is a daunting task, and the traditional methods used for this virtual "screening" process have their limitations.
Recently, scientists have started using a new technique called deep generative modeling, which essentially teaches an AI system to generate new molecules based on existing data. The idea is that this AI can explore the chemical space more efficiently than traditional methods. However, these models tend to get stuck generating molecules that are similar to those in their training data, rather than coming up with truly novel and improved compounds.
The researchers in this paper propose a different approach, where they frame the molecule generation task as a reinforcement learning problem. Instead of just trying to mimic the training data, the AI is given specific goals to optimize for, like having strong binding affinity to the target protein, being "drug-like" (having properties similar to existing medications), and being relatively easy to synthesize in a laboratory. By setting these objectives and letting the AI explore more freely, the researchers were able to generate new molecules that outperformed state-of-the-art methods on several key metrics.
Technical Explanation
The central idea of this paper is to frame the task of generating novel, high-performing drug molecules as a reinforcement learning (RL) problem, rather than a standard generative modeling approach.
The researchers developed a model called TacoGFN, which is a Generative Flow Network that is conditioned on the structure of the protein target. This allows the model to take the 3D shape of the protein binding pocket into account when generating new molecules.
The key innovation is that TacoGFN is trained using RL, with the goal of maximizing a reward function that combines three important molecular properties: binding affinity to the target protein (as measured by docking score), drug-likeness (using the Quantitative Estimation of Druglikeness, or QED metric), and synthetic accessibility (using the Synthetic Accessibility score, or SA).
By optimizing directly for these desirable characteristics, rather than just trying to match a training data distribution, the model is able to explore the wider chemical space and generate novel molecules that significantly outperform state-of-the-art methods on the CrossDocked2020 benchmark. Specifically, TacoGFN achieved a median docking score of -8.82 and a 52.63% novel hit rate.
Critical Analysis
The authors acknowledge that their approach has some limitations. For example, the reliance on docking scores as a proxy for binding affinity may not fully capture the complex dynamics of protein-ligand interactions. Additionally, the reinforcement learning setup means the model's performance is heavily dependent on the choice and weighting of the reward function components.
Furthermore, the paper does not provide a detailed analysis of the chemical diversity or novelty of the generated molecules beyond the "novel hit rate" metric. It would be valuable to see a more in-depth characterization of the structural and property landscapes explored by TacoGFN.
That said, the overall results are impressive, and the reinforcement learning framing is a clever way to push generative models beyond the confines of their training data distribution. Further research in this direction, with more comprehensive evaluations, could yield significant advancements in the field of computational drug discovery.
Conclusion
This paper presents a novel approach to the challenge of generating drug-like and synthesizable molecules with high binding affinity to target proteins. By formulating the task as a reinforcement learning problem, the researchers were able to develop a model (TacoGFN) that outperformed state-of-the-art methods on several key metrics.
The ability to explore the wider chemical space and optimize directly for desirable molecular properties, rather than just fitting a training data distribution, is a significant advancement in the field of computational drug discovery. While the approach has some limitations, the strong results suggest that this reinforcement learning framework could lead to the identification of promising drug candidates more efficiently than traditional virtual screening methods.
As this research progresses, it will be important to further explore the chemical diversity and novelty of the generated molecules, as well as to validate the findings through experimental testing. Nevertheless, this work represents an important step forward in the quest to harness the power of AI and machine learning for accelerating the drug discovery process.
This summary was produced with help from an AI and may contain inaccuracies - check out the links to read the original source documents!
Related Papers

Geometric-informed GFlowNets for Structure-Based Drug Design
Grayson Lee, Tony Shen, Martin Ester
0
0
The rise of cost involved with drug discovery and current speed of which they are discover, underscore the need for more efficient structure-based drug design (SBDD) methods. We employ Generative Flow Networks (GFlowNets), to effectively explore the vast combinatorial space of drug-like molecules, which traditional virtual screening methods fail to cover. We introduce a novel modification to the GFlowNet framework by incorporating trigonometrically consistent embeddings, previously utilized in tasks involving protein conformation and protein-ligand interactions, to enhance the model's ability to generate molecules tailored to specific protein pockets. We have modified the existing protein conditioning used by GFlowNets, blending geometric information from both protein and ligand embeddings to achieve more geometrically consistent embeddings. Experiments conducted using CrossDocked2020 demonstrated an improvement in the binding affinity between generated molecules and protein pockets for both single and multi-objective tasks, compared to previous work. Additionally, we propose future work aimed at further increasing the geometric information captured in protein-ligand interactions.
6/18/2024
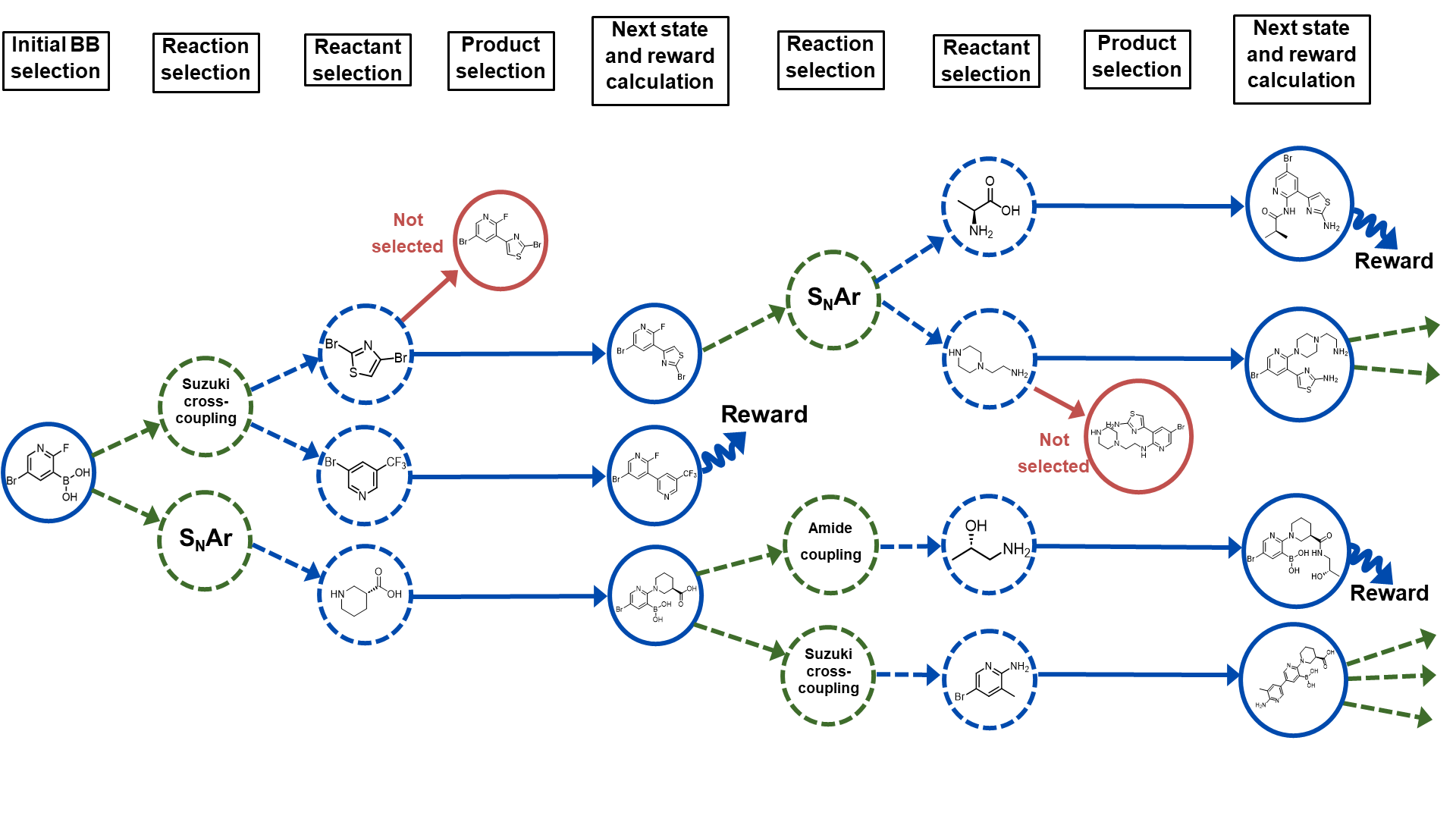
RGFN: Synthesizable Molecular Generation Using GFlowNets
Micha{l} Koziarski, Andrei Rekesh, Dmytro Shevchuk, Almer van der Sloot, Piotr Gai'nski, Yoshua Bengio, Cheng-Hao Liu, Mike Tyers, Robert A. Batey
0
0
Generative models hold great promise for small molecule discovery, significantly increasing the size of search space compared to traditional in silico screening libraries. However, most existing machine learning methods for small molecule generation suffer from poor synthesizability of candidate compounds, making experimental validation difficult. In this paper we propose Reaction-GFlowNet (RGFN), an extension of the GFlowNet framework that operates directly in the space of chemical reactions, thereby allowing out-of-the-box synthesizability while maintaining comparable quality of generated candidates. We demonstrate that with the proposed set of reactions and building blocks, it is possible to obtain a search space of molecules orders of magnitude larger than existing screening libraries coupled with low cost of synthesis. We also show that the approach scales to very large fragment libraries, further increasing the number of potential molecules. We demonstrate the effectiveness of the proposed approach across a range of oracle models, including pretrained proxy models and GPU-accelerated docking.
6/14/2024
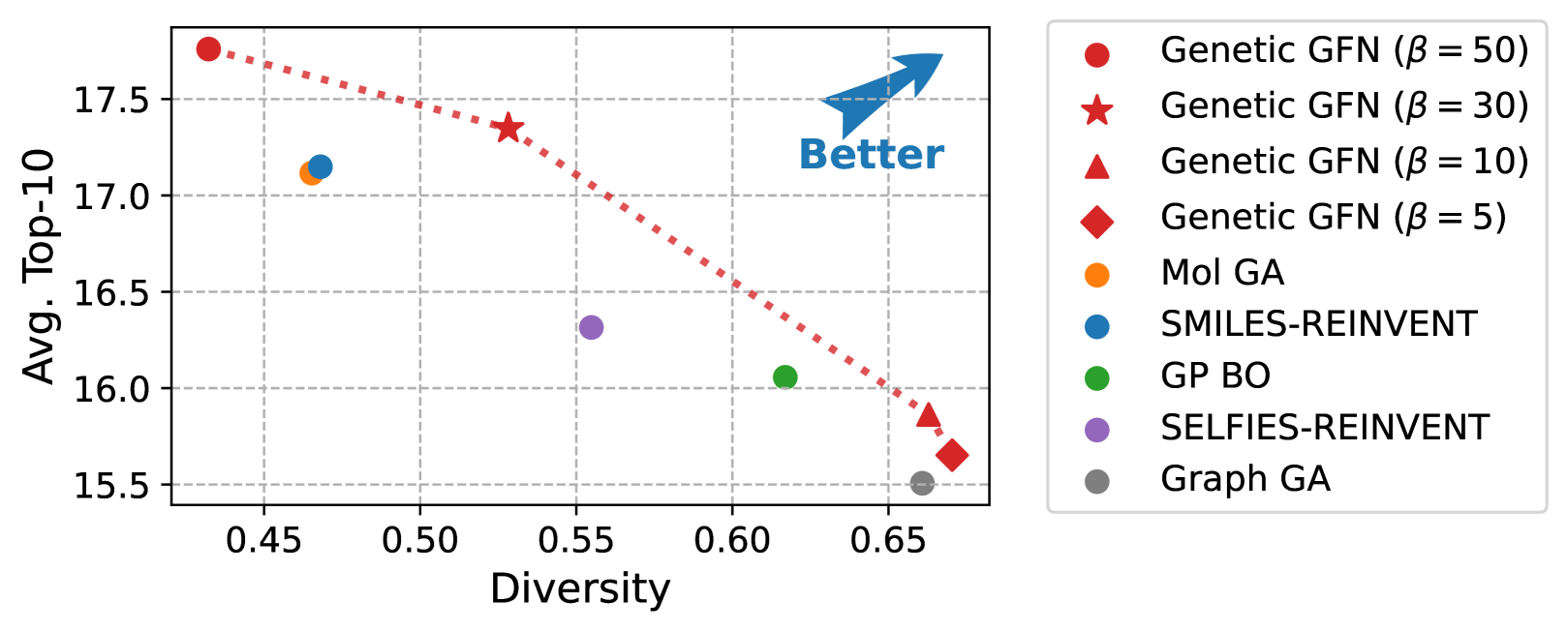
Genetic-guided GFlowNets for Sample Efficient Molecular Optimization
Hyeonah Kim, Minsu Kim, Sanghyeok Choi, Jinkyoo Park
0
0
The challenge of discovering new molecules with desired properties is crucial in domains like drug discovery and material design. Recent advances in deep learning-based generative methods have shown promise but face the issue of sample efficiency due to the computational expense of evaluating the reward function. This paper proposes a novel algorithm for sample-efficient molecular optimization by distilling a powerful genetic algorithm into deep generative policy using GFlowNets training, the off-policy method for amortized inference. This approach enables the deep generative policy to learn from domain knowledge, which has been explicitly integrated into the genetic algorithm. Our method achieves state-of-the-art performance in the official molecular optimization benchmark, significantly outperforming previous methods. It also demonstrates effectiveness in designing inhibitors against SARS-CoV-2 with substantially fewer reward calls.
5/28/2024

SynFlowNet: Towards Molecule Design with Guaranteed Synthesis Pathways
Miruna Cretu, Charles Harris, Julien Roy, Emmanuel Bengio, Pietro Li`o
0
0
Recent breakthroughs in generative modelling have led to a number of works proposing molecular generation models for drug discovery. While these models perform well at capturing drug-like motifs, they are known to often produce synthetically inaccessible molecules. This is because they are trained to compose atoms or fragments in a way that approximates the training distribution, but they are not explicitly aware of the synthesis constraints that come with making molecules in the lab. To address this issue, we introduce SynFlowNet, a GFlowNet model whose action space uses chemically validated reactions and reactants to sequentially build new molecules. We evaluate our approach using synthetic accessibility scores and an independent retrosynthesis tool. SynFlowNet consistently samples synthetically feasible molecules, while still being able to find diverse and high-utility candidates. Furthermore, we compare molecules designed with SynFlowNet to experimentally validated actives, and find that they show comparable properties of interest, such as molecular weight, SA score and predicted protein binding affinity.
5/3/2024