On the Scalability of GNNs for Molecular Graphs
2404.11568
0
0
Abstract
Scaling deep learning models has been at the heart of recent revolutions in language modelling and image generation. Practitioners have observed a strong relationship between model size, dataset size, and performance. However, structure-based architectures such as Graph Neural Networks (GNNs) are yet to show the benefits of scale mainly due to the lower efficiency of sparse operations, large data requirements, and lack of clarity about the effectiveness of various architectures. We address this drawback of GNNs by studying their scaling behavior. Specifically, we analyze message-passing networks, graph Transformers, and hybrid architectures on the largest public collection of 2D molecular graphs. For the first time, we observe that GNNs benefit tremendously from the increasing scale of depth, width, number of molecules, number of labels, and the diversity in the pretraining datasets. We further demonstrate strong finetuning scaling behavior on 38 highly competitive downstream tasks, outclassing previous large models. This gives rise to MolGPS, a new graph foundation model that allows to navigate the chemical space, outperforming the previous state-of-the-arts on 26 out the 38 downstream tasks. We hope that our work paves the way for an era where foundational GNNs drive pharmaceutical drug discovery.
Create account to get full access
Overview
- This paper explores the scalability of Graph Neural Networks (GNNs) for molecular graph datasets.
- The authors investigate the computational and memory costs of GNNs as the size of molecular graphs increases.
- They propose an approach to improve the scalability of GNNs for large molecular graphs.
Plain English Explanation
Graph Neural Networks (GNNs) are a type of machine learning model that can process data represented as graphs, such as molecular structures. Molecular graphs are an important application of GNNs, as they can be used to predict properties of chemical compounds.
However, as the size of molecular graphs increases, the computational and memory requirements of GNNs can become a challenge. This paper examines this issue and proposes a solution to improve the scalability of GNNs for large molecular graphs.
The authors investigate the performance of GNNs on larger molecular datasets, measuring factors like runtime and memory usage. They find that the costs can grow rapidly as the graphs get bigger. To address this, they develop a new approach that can handle larger molecular graphs more efficiently.
This research is significant because it helps make GNNs more practical for real-world applications involving large, complex molecular structures. By improving the scalability of these models, the authors pave the way for more accurate and efficient predictions of chemical properties, which could have important implications for drug discovery, materials science, and other fields.
Technical Explanation
The paper starts by reviewing related work on the use of GNNs for molecular modeling. It then provides background on the key concepts underlying GNNs and their application to molecular graphs.
The main focus of the paper is an empirical analysis of the scalability of GNNs for large molecular datasets. The authors measure the computational runtime and memory requirements of several GNN architectures as the size of the molecular graphs increases.
Their results show that the costs can grow rapidly, making GNNs impractical for very large molecular datasets. To address this, the authors propose a novel GNN architecture that is more scalable. This new model uses techniques like graph coarsening and sparsity-aware operators to reduce the computational burden.
The paper includes extensive experiments evaluating the performance of the proposed approach on a range of molecular benchmarks. The results demonstrate significant improvements in runtime and memory usage compared to standard GNN models, especially for the largest molecular graphs.
Critical Analysis
The paper provides a thorough and well-designed analysis of the scalability challenges faced by GNNs in the context of molecular modeling. The authors carefully consider the computational and memory costs of GNNs as the graph size increases, and their proposed solution appears to be a promising approach to address these issues.
However, the paper does not delve into some potential limitations or areas for further research. For example, the authors do not discuss how their approach might perform on even larger or more complex molecular datasets beyond those included in the experiments. Additionally, there may be other techniques or architectural innovations that could further improve the scalability of GNNs for this domain.
It would also be valuable to see the authors contextualize their findings more broadly, such as how this work relates to the development of large language models and their implications for the field of graph machine learning.
Overall, this paper makes a significant contribution to improving the scalability of GNNs for molecular applications, but there may be opportunities for further research and refinement of the proposed approach.
Conclusion
This paper tackles an important challenge in the application of Graph Neural Networks (GNNs) to molecular graph datasets – the issue of scalability as the size of the graphs increases. The authors provide a thorough empirical analysis of the computational and memory costs of GNNs, and they propose a novel GNN architecture that demonstrates significant improvements in scalability.
This research is valuable because it helps address a key barrier to the wider adoption of GNNs in real-world molecular modeling applications, which often involve large and complex chemical structures. By making GNNs more scalable, the authors pave the way for more accurate and efficient predictions of chemical properties, which could have important implications for drug discovery, materials science, and other fields.
While the paper does not explore all possible avenues for further improving GNN scalability, it represents an important step forward in this area of research. The insights and techniques developed in this work could also be applicable to other graph-based domains beyond molecular modeling, contributing to the overall advancement of graph machine learning.
This summary was produced with help from an AI and may contain inaccuracies - check out the links to read the original source documents!
Related Papers

Scalable Training of Graph Foundation Models for Atomistic Materials Modeling: A Case Study with HydraGNN
Massimiliano Lupo Pasini, Jong Youl Choi, Kshitij Mehta, Pei Zhang, David Rogers, Jonghyun Bae, Khaled Z. Ibrahim, Ashwin M. Aji, Karl W. Schulz, Jorda Polo, Prasanna Balaprakash
0
0
We present our work on developing and training scalable graph foundation models (GFM) using HydraGNN, a multi-headed graph convolutional neural network architecture. HydraGNN expands the boundaries of graph neural network (GNN) in both training scale and data diversity. It abstracts over message passing algorithms, allowing both reproduction of and comparison across algorithmic innovations that define convolution in GNNs. This work discusses a series of optimizations that have allowed scaling up the GFM training to tens of thousands of GPUs on datasets that consist of hundreds of millions of graphs. Our GFMs use multi-task learning (MTL) to simultaneously learn graph-level and node-level properties of atomistic structures, such as the total energy and atomic forces. Using over 150 million atomistic structures for training, we illustrate the performance of our approach along with the lessons learned on two United States Department of Energy (US-DOE) supercomputers, namely the Perlmutter petascale system at the National Energy Research Scientific Computing Center and the Frontier exascale system at Oak Ridge National Laboratory. The HydraGNN architecture enables the GFM to achieve near-linear strong scaling performance using more than 2,000 GPUs on Perlmutter and 16,000 GPUs on Frontier. Hyperparameter optimization (HPO) was performed on over 64,000 GPUs on Frontier to select GFM architectures with high accuracy. Early stopping was applied on each GFM architecture for energy awareness in performing such an extreme-scale task. The training of an ensemble of highest-ranked GFM architectures continued until convergence to establish uncertainty quantification (UQ) capabilities with ensemble learning. Our contribution opens the door for rapidly developing, training, and deploying GFMs using large-scale computational resources to enable AI-accelerated materials discovery and design.
7/1/2024
A survey of dynamic graph neural networks
Yanping Zheng, Lu Yi, Zhewei Wei
0
0
Graph neural networks (GNNs) have emerged as a powerful tool for effectively mining and learning from graph-structured data, with applications spanning numerous domains. However, most research focuses on static graphs, neglecting the dynamic nature of real-world networks where topologies and attributes evolve over time. By integrating sequence modeling modules into traditional GNN architectures, dynamic GNNs aim to bridge this gap, capturing the inherent temporal dependencies of dynamic graphs for a more authentic depiction of complex networks. This paper provides a comprehensive review of the fundamental concepts, key techniques, and state-of-the-art dynamic GNN models. We present the mainstream dynamic GNN models in detail and categorize models based on how temporal information is incorporated. We also discuss large-scale dynamic GNNs and pre-training techniques. Although dynamic GNNs have shown superior performance, challenges remain in scalability, handling heterogeneous information, and lack of diverse graph datasets. The paper also discusses possible future directions, such as adaptive and memory-enhanced models, inductive learning, and theoretical analysis.
4/30/2024
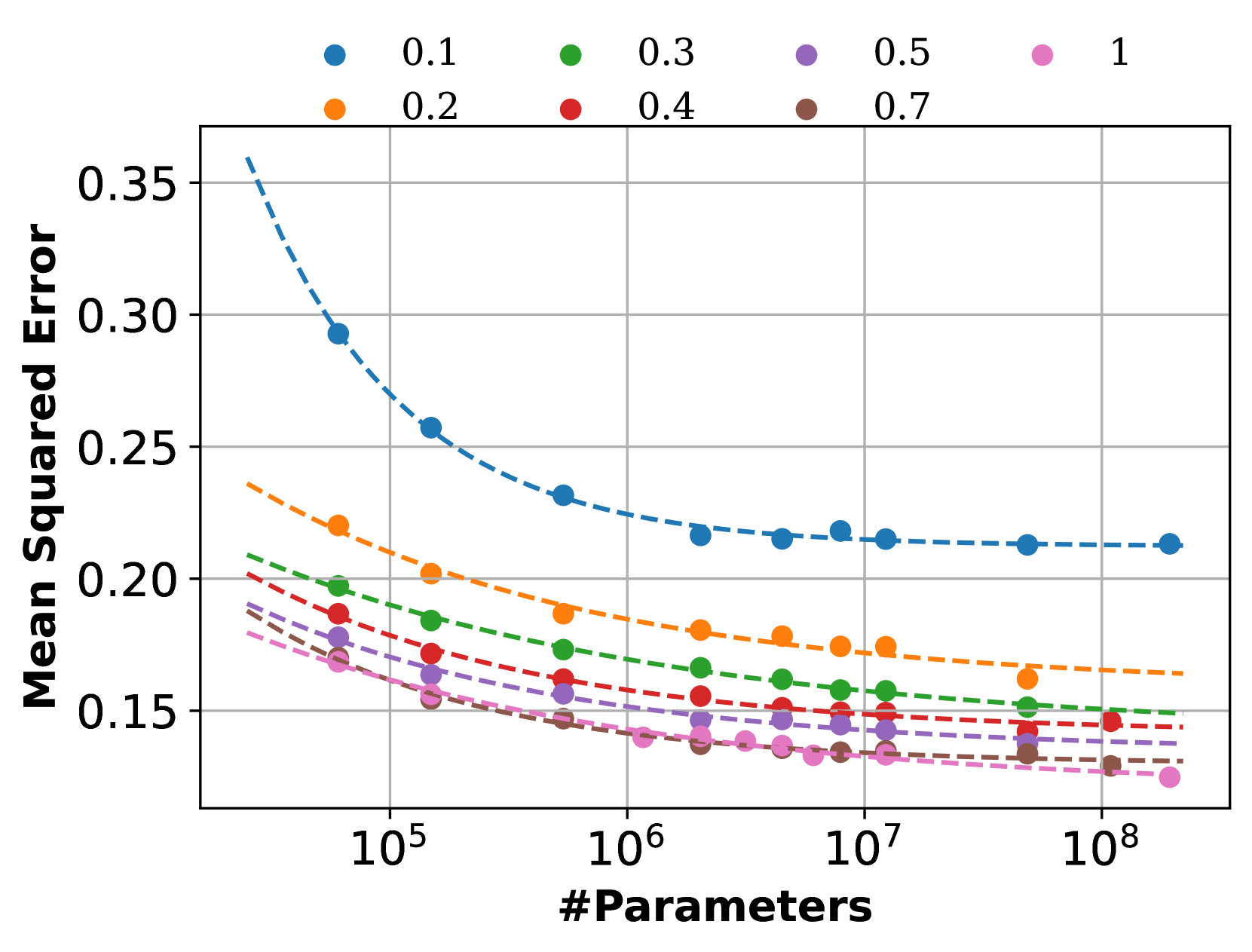
Neural Scaling Laws on Graphs
Jingzhe Liu, Haitao Mao, Zhikai Chen, Tong Zhao, Neil Shah, Jiliang Tang
0
0
Deep graph models (e.g., graph neural networks and graph transformers) have become important techniques for leveraging knowledge across various types of graphs. Yet, the scaling properties of deep graph models have not been systematically investigated, casting doubt on the feasibility of achieving large graph models through enlarging the model and dataset sizes. In this work, we delve into neural scaling laws on graphs from both model and data perspectives. We first verify the validity of such laws on graphs, establishing formulations to describe the scaling behaviors. For model scaling, we investigate the phenomenon of scaling law collapse and identify overfitting as the potential reason. Moreover, we reveal that the model depth of deep graph models can impact the model scaling behaviors, which differ from observations in other domains such as CV and NLP. For data scaling, we suggest that the number of graphs can not effectively metric the graph data volume in scaling law since the sizes of different graphs are highly irregular. Instead, we reform the data scaling law with the number of edges as the metric to address the irregular graph sizes. We further demonstrate the reformed law offers a unified view of the data scaling behaviors for various fundamental graph tasks including node classification, link prediction, and graph classification. This work provides valuable insights into neural scaling laws on graphs, which can serve as an essential step toward large graph models.
6/11/2024
🤿
Lightweight Geometric Deep Learning for Molecular Modelling in Catalyst Discovery
Patrick Geitner
0
0
New technology for energy storage is necessary for the large-scale adoption of renewable energy sources like wind and solar. The ability to discover suitable catalysts is crucial for making energy storage more cost-effective and scalable. The Open Catalyst Project aims to apply advances in graph neural networks (GNNs) to accelerate progress in catalyst discovery, replacing Density Functional Theory-based (DFT) approaches that are computationally burdensome. Current approaches involve scaling GNNs to over 1 billion parameters, pushing the problem out of reach for a vast majority of machine learning practitioner around the world. This study aims to evaluate the performance and insights gained from using more lightweight approaches for this task that are more approachable for smaller teams to encourage participation from individuals from diverse backgrounds. By implementing robust design patterns like geometric and symmetric message passing, we were able to train a GNN model that reached a MAE of 0.0748 in predicting the per-atom forces of adsorbate-surface interactions, rivaling established model architectures like SchNet and DimeNet++ while using only a fraction of trainable parameters.
4/17/2024