Scalable Training of Graph Foundation Models for Atomistic Materials Modeling: A Case Study with HydraGNN

0

Sign in to get full access
Overview
- This research paper explores scalable training of graph foundation models for atomistic materials modeling, using a case study with HydraGNN.
- It is sponsored by the Artificial Intelligence Initiative as part of the Laboratory Directed Research and Development (LDRD) Program of Oak Ridge National Laboratory.
- The work utilized resources from the Oak Ridge Leadership Computing Facility, supported by the U.S. Department of Energy.
Plain English Explanation
This paper describes how the researchers developed a way to efficiently train large-scale graph neural network models, which are a type of machine learning algorithm, to help with understanding the properties of materials at the atomic level. Graph neural networks are a powerful tool for analyzing the connections and relationships in data that can be represented as a graph, such as the structure of molecules and materials.
The researchers used a technique called "data parallelism" to split up the training of the model across multiple computers, allowing them to handle much larger and more complex datasets than would be possible on a single machine. This is an important advance, as graph foundation models are increasingly being used to model the behavior of materials at the atomic scale, but require vast amounts of data and computational power to train effectively.
By scaling up the training of these graph neural network models, the researchers have made it possible to apply them to real-world materials science problems, such as designing new catalysts or predicting the properties of molecules. This could lead to faster and more efficient development of new materials with desirable properties, ultimately benefiting fields like clean energy, medicine, and more.
Technical Explanation
The key technical contribution of this paper is the development of a scalable training approach for graph foundation models, demonstrated through a case study with the HydraGNN architecture. The researchers utilized data parallelism, a technique in which the training data is divided across multiple GPU devices, to enable the efficient training of large-scale graph neural network models.
The HydraGNN architecture is a message-passing graph neural network that can learn representations of atomistic materials data. The researchers showed that by employing data parallelism, they were able to scale the training of HydraGNN to handle much larger and more complex datasets than would be possible on a single GPU. This was achieved by partitioning the training data and gradients across multiple devices, allowing the model to be trained in a distributed and parallel fashion.
The researchers conducted extensive experiments to evaluate the scalability and performance of their approach, demonstrating its ability to effectively train HydraGNN on large-scale materials science datasets. They compared the performance of the data-parallel training to that of single-GPU training, showcasing significant improvements in training speed and model performance.
Critical Analysis
The researchers have presented a compelling approach to scaling the training of graph foundation models for atomistic materials modeling. By leveraging data parallelism, they were able to overcome the computational limitations of training these models on a single GPU, a critical barrier to their wider adoption in materials science research.
One potential caveat is that the paper focuses solely on the HydraGNN architecture and does not explore the scalability of other graph neural network models. While HydraGNN is a well-established and effective architecture, it would be valuable to see if the data-parallel training approach can be generalized to other graph foundation models as well.
Additionally, the paper does not delve into the practical considerations of deploying such a scalable training system in a real-world research or production environment. Further work may be needed to address issues like fault tolerance, monitoring, and integration with existing materials science workflows.
Overall, this research represents an important step forward in making graph foundation models more accessible and scalable for atomistic materials modeling. By reducing the computational barriers to training these models, the researchers have opened up new possibilities for their application in materials discovery, optimization, and simulation.
Conclusion
This paper presents a significant advancement in the scalable training of graph foundation models for atomistic materials modeling. By employing data parallelism, the researchers were able to overcome the limitations of single-GPU training and enable the efficient training of large-scale graph neural network models, such as HydraGNN.
The ability to scale the training of these models is a crucial step in their broader adoption and application in materials science research. With the capacity to handle larger and more complex datasets, researchers can now leverage the power of graph foundation models to gain deeper insights into the behavior of materials at the atomic level, accelerating the development of new materials with desirable properties.
The techniques and insights presented in this paper have the potential to benefit a wide range of fields, from clean energy and medicine to manufacturing and beyond. As graph foundation models continue to evolve and become more accessible, the impact of this research on materials science and beyond could be far-reaching.
This summary was produced with help from an AI and may contain inaccuracies - check out the links to read the original source documents!
Related Papers
0
Scalable Training of Graph Foundation Models for Atomistic Materials Modeling: A Case Study with HydraGNN
Massimiliano Lupo Pasini, Jong Youl Choi, Kshitij Mehta, Pei Zhang, David Rogers, Jonghyun Bae, Khaled Z. Ibrahim, Ashwin M. Aji, Karl W. Schulz, Jorda Polo, Prasanna Balaprakash
We present our work on developing and training scalable graph foundation models (GFM) using HydraGNN, a multi-headed graph convolutional neural network architecture. HydraGNN expands the boundaries of graph neural network (GNN) in both training scale and data diversity. It abstracts over message passing algorithms, allowing both reproduction of and comparison across algorithmic innovations that define convolution in GNNs. This work discusses a series of optimizations that have allowed scaling up the GFM training to tens of thousands of GPUs on datasets that consist of hundreds of millions of graphs. Our GFMs use multi-task learning (MTL) to simultaneously learn graph-level and node-level properties of atomistic structures, such as the total energy and atomic forces. Using over 150 million atomistic structures for training, we illustrate the performance of our approach along with the lessons learned on two United States Department of Energy (US-DOE) supercomputers, namely the Perlmutter petascale system at the National Energy Research Scientific Computing Center and the Frontier exascale system at Oak Ridge National Laboratory. The HydraGNN architecture enables the GFM to achieve near-linear strong scaling performance using more than 2,000 GPUs on Perlmutter and 16,000 GPUs on Frontier. Hyperparameter optimization (HPO) was performed on over 64,000 GPUs on Frontier to select GFM architectures with high accuracy. Early stopping was applied on each GFM architecture for energy awareness in performing such an extreme-scale task. The training of an ensemble of highest-ranked GFM architectures continued until convergence to establish uncertainty quantification (UQ) capabilities with ensemble learning. Our contribution opens the door for rapidly developing, training, and deploying GFMs using large-scale computational resources to enable AI-accelerated materials discovery and design.
Read more7/1/2024
0
On the Scalability of GNNs for Molecular Graphs
Maciej Sypetkowski, Frederik Wenkel, Farimah Poursafaei, Nia Dickson, Karush Suri, Philip Fradkin, Dominique Beaini
Scaling deep learning models has been at the heart of recent revolutions in language modelling and image generation. Practitioners have observed a strong relationship between model size, dataset size, and performance. However, structure-based architectures such as Graph Neural Networks (GNNs) are yet to show the benefits of scale mainly due to the lower efficiency of sparse operations, large data requirements, and lack of clarity about the effectiveness of various architectures. We address this drawback of GNNs by studying their scaling behavior. Specifically, we analyze message-passing networks, graph Transformers, and hybrid architectures on the largest public collection of 2D molecular graphs. For the first time, we observe that GNNs benefit tremendously from the increasing scale of depth, width, number of molecules, number of labels, and the diversity in the pretraining datasets. We further demonstrate strong finetuning scaling behavior on 38 highly competitive downstream tasks, outclassing previous large models. This gives rise to MolGPS, a new graph foundation model that allows to navigate the chemical space, outperforming the previous state-of-the-arts on 26 out the 38 downstream tasks. We hope that our work paves the way for an era where foundational GNNs drive pharmaceutical drug discovery.
Read more9/12/2024

0
GraphFM: A Scalable Framework for Multi-Graph Pretraining
Divyansha Lachi, Mehdi Azabou, Vinam Arora, Eva Dyer
Graph neural networks are typically trained on individual datasets, often requiring highly specialized models and extensive hyperparameter tuning. This dataset-specific approach arises because each graph dataset often has unique node features and diverse connectivity structures, making it difficult to build a generalist model. To address these challenges, we introduce a scalable multi-graph multi-task pretraining approach specifically tailored for node classification tasks across diverse graph datasets from different domains. Our method, Graph Foundation Model (GraphFM), leverages a Perceiver-based encoder that employs learned latent tokens to compress domain-specific features into a common latent space. This approach enhances the model's ability to generalize across different graphs and allows for scaling across diverse data. We demonstrate the efficacy of our approach by training a model on 152 different graph datasets comprising over 7.4 million nodes and 189 million edges, establishing the first set of scaling laws for multi-graph pretraining on datasets spanning many domains (e.g., molecules, citation and product graphs). Our results show that pretraining on a diverse array of real and synthetic graphs improves the model's adaptability and stability, while performing competitively with state-of-the-art specialist models. This work illustrates that multi-graph pretraining can significantly reduce the burden imposed by the current graph training paradigm, unlocking new capabilities for the field of graph neural networks by creating a single generalist model that performs competitively across a wide range of datasets and tasks.
Read more7/17/2024
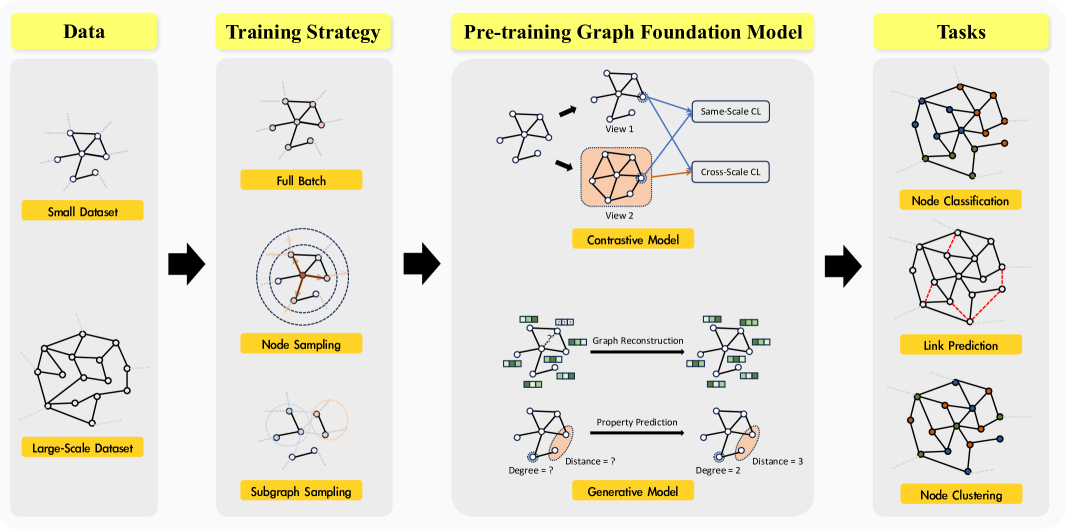
0
GraphFM: A Comprehensive Benchmark for Graph Foundation Model
Yuhao Xu, Xinqi Liu, Keyu Duan, Yi Fang, Yu-Neng Chuang, Daochen Zha, Qiaoyu Tan
Foundation Models (FMs) serve as a general class for the development of artificial intelligence systems, offering broad potential for generalization across a spectrum of downstream tasks. Despite extensive research into self-supervised learning as the cornerstone of FMs, several outstanding issues persist in Graph Foundation Models that rely on graph self-supervised learning, namely: 1) Homogenization. The extent of generalization capability on downstream tasks remains unclear. 2) Scalability. It is unknown how effectively these models can scale to large datasets. 3) Efficiency. The training time and memory usage of these models require evaluation. 4) Training Stop Criteria. Determining the optimal stopping strategy for pre-training across multiple tasks to maximize performance on downstream tasks. To address these questions, we have constructed a rigorous benchmark that thoroughly analyzes and studies the generalization and scalability of self-supervised Graph Neural Network (GNN) models. Regarding generalization, we have implemented and compared the performance of various self-supervised GNN models, trained to generate node representations, across tasks such as node classification, link prediction, and node clustering. For scalability, we have compared the performance of various models after training using full-batch and mini-batch strategies. Additionally, we have assessed the training efficiency of these models by conducting experiments to test their GPU memory usage and throughput. Through these experiments, we aim to provide insights to motivate future research. The code for this benchmark is publicly available at https://github.com/NYUSHCS/GraphFM.
Read more6/17/2024