A Self-feedback Knowledge Elicitation Approach for Chemical Reaction Predictions

0
Sign in to get full access
Overview
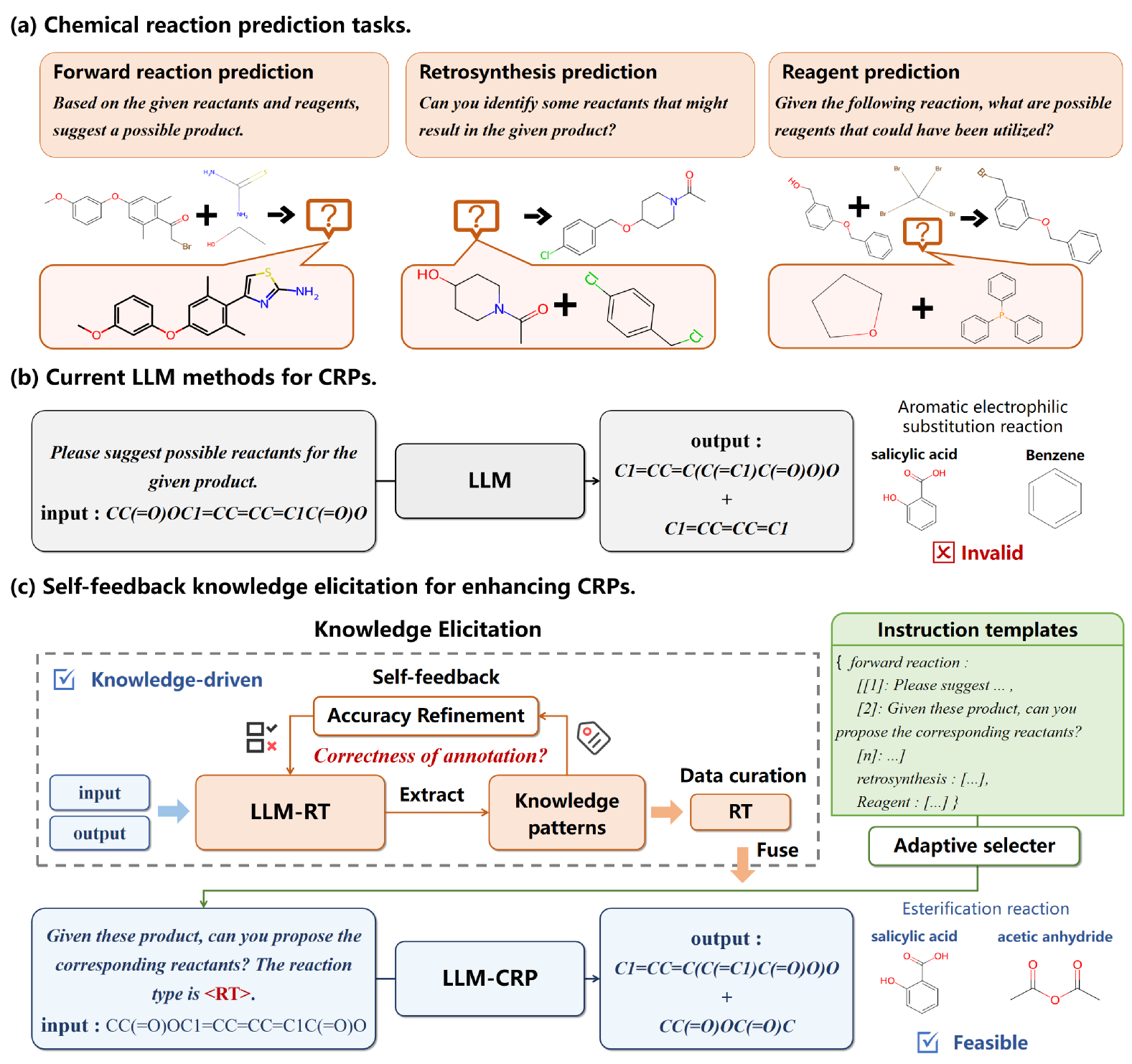
- This paper presents a self-feedback knowledge elicitation approach for improving the performance of large language models on chemical reaction predictions.
- The method involves iteratively prompting the model with its own outputs, allowing it to refine and improve its understanding of chemical reaction patterns over time.
- The authors demonstrate the effectiveness of this approach on a benchmark chemical reaction dataset, showing significant improvements in predictive accuracy compared to standard fine-tuning methods.
Plain English Explanation
Large language models like the ones discussed in this paper are powerful tools for tasks like chemical reaction prediction, but they can struggle to fully capture the complex underlying patterns. This paper proposes a novel approach to address this challenge.
The key idea is to let the language model learn from its own outputs. Instead of just fine-tuning the model on a dataset, the researchers iteratively prompt the model with its own previous responses. This allows the model to refine and deepen its understanding of chemical reaction dynamics over time.
Imagine you're a student learning about chemistry. At first, you might struggle to fully grasp all the nuances of how different compounds react. But if you're given a problem, try to solve it, and then get feedback on your answer, you can use that information to improve your knowledge for the next problem. That's essentially what this self-feedback approach does for the language model.
By repeatedly exposing the model to its own outputs and allowing it to learn from them, the researchers were able to significantly boost the model's predictive accuracy on a benchmark chemical reaction dataset. This suggests that this self-feedback technique could be a powerful way to help large language models become more effective at understanding and reasoning about complex scientific domains like chemistry.
Technical Explanation
The core of this paper's approach is a self-feedback knowledge elicitation technique for improving large language model performance on chemical reaction prediction tasks. The authors start with a pre-trained language model and fine-tune it on a dataset of chemical reactions using standard methods.
However, they then go a step further by iteratively prompting the model with its own previous outputs. Specifically, they take the model's predictions for a given input, package those predictions back into a new prompt, and feed that prompt back into the model. This allows the model to refine and expand its understanding of chemical reaction patterns over multiple rounds of self-feedback.
The authors evaluate this approach on the USPTO chemical reaction dataset and demonstrate significant improvements in predictive accuracy compared to standard fine-tuning. They also conduct ablation studies to show the importance of the self-feedback component, as opposed to simply increasing the amount of fine-tuning data.
One key insight from the paper is that this self-feedback process helps the model better capture long-range dependencies and complex reaction mechanisms that may be difficult to learn from the training data alone. By iteratively refining its outputs, the model is able to develop a more holistic understanding of the underlying chemical principles.
Critical Analysis
The authors present a compelling approach for enhancing large language model performance on chemical reaction prediction tasks. The self-feedback technique is a novel and promising idea that builds on recent advances in prompt learning and active learning.
That said, the paper does not fully address the potential limitations of this approach. For example, the self-feedback process could potentially amplify biases or errors in the model's initial outputs, leading to suboptimal refinement over successive iterations. The authors also do not discuss how the self-feedback technique might scale to larger and more diverse chemical reaction datasets.
Additionally, while the results on the USPTO benchmark are impressive, it would be valuable to see how the self-feedback approach performs on other chemical prediction tasks or real-world applications. Encouraging readers to think critically about the research and its broader implications is an important part of a balanced technical summary.
Conclusion
This paper presents a novel self-feedback knowledge elicitation technique that significantly improves the performance of large language models on chemical reaction prediction tasks. By iteratively prompting the model with its own outputs, the approach allows the model to refine and deepen its understanding of complex chemical reaction patterns.
The results demonstrate the potential of this approach to enhance the capabilities of language models in important scientific domains like chemistry. As large language models continue to advance, techniques like self-feedback could play a key role in unlocking their full potential for complex reasoning and problem-solving. This work represents an important step forward in this direction.
This summary was produced with help from an AI and may contain inaccuracies - check out the links to read the original source documents!
Related Papers
0
A Self-feedback Knowledge Elicitation Approach for Chemical Reaction Predictions
Pengfei Liu, Jun Tao, Zhixiang Ren
The task of chemical reaction predictions (CRPs) plays a pivotal role in advancing drug discovery and material science. However, its effectiveness is constrained by the vast and uncertain chemical reaction space and challenges in capturing reaction selectivity, particularly due to existing methods' limitations in exploiting the data's inherent knowledge. To address these challenges, we introduce a data-curated self-feedback knowledge elicitation approach. This method starts from iterative optimization of molecular representations and facilitates the extraction of knowledge on chemical reaction types (RTs). Then, we employ adaptive prompt learning to infuse the prior knowledge into the large language model (LLM). As a result, we achieve significant enhancements: a 14.2% increase in retrosynthesis prediction accuracy, a 74.2% rise in reagent prediction accuracy, and an expansion in the model's capability for handling multi-task chemical reactions. This research offers a novel paradigm for knowledge elicitation in scientific research and showcases the untapped potential of LLMs in CRPs.
Read more4/16/2024

0
ChemReasoner: Heuristic Search over a Large Language Model's Knowledge Space using Quantum-Chemical Feedback
Henry W. Sprueill, Carl Edwards, Khushbu Agarwal, Mariefel V. Olarte, Udishnu Sanyal, Conrad Johnston, Hongbin Liu, Heng Ji, Sutanay Choudhury
The discovery of new catalysts is essential for the design of new and more efficient chemical processes in order to transition to a sustainable future. We introduce an AI-guided computational screening framework unifying linguistic reasoning with quantum-chemistry based feedback from 3D atomistic representations. Our approach formulates catalyst discovery as an uncertain environment where an agent actively searches for highly effective catalysts via the iterative combination of large language model (LLM)-derived hypotheses and atomistic graph neural network (GNN)-derived feedback. Identified catalysts in intermediate search steps undergo structural evaluation based on spatial orientation, reaction pathways, and stability. Scoring functions based on adsorption energies and reaction energy barriers steer the exploration in the LLM's knowledge space toward energetically favorable, high-efficiency catalysts. We introduce planning methods that automatically guide the exploration without human input, providing competitive performance against expert-enumerated chemical descriptor-based implementations. By integrating language-guided reasoning with computational chemistry feedback, our work pioneers AI-accelerated, trustworthy catalyst discovery.
Read more6/10/2024

0
Chemical Reaction Extraction for Chemical Knowledge Base
Aishwarya Jadhav, Ritam Dutt
The task of searching through patent documents is crucial for chemical patent recommendation and retrieval. This can be enhanced by creating a patent knowledge base (ChemPatKB) to aid in prior art searches and to provide a platform for domain experts to explore new innovations in chemical compound synthesis and use-cases. An essential foundational component of this KB is the extraction of important reaction snippets from long patents documents which facilitates multiple downstream tasks such as reaction co-reference resolution and chemical entity role identification. In this work, we explore the problem of extracting reactions spans from chemical patents in order to create a reactions resource database. We formulate this task as a paragraph-level sequence tagging problem, where the system is required to return a sequence of paragraphs that contain a description of a reaction. We propose several approaches and modifications of the baseline models and study how different methods generalize across different domains of chemical patents.
Read more7/24/2024
💬
0
Chemist-X: Large Language Model-empowered Agent for Reaction Condition Recommendation in Chemical Synthesis
Kexin Chen, Junyou Li, Kunyi Wang, Yuyang Du, Jiahui Yu, Jiamin Lu, Lanqing Li, Jiezhong Qiu, Jianzhang Pan, Yi Huang, Qun Fang, Pheng Ann Heng, Guangyong Chen
Recent AI research plots a promising future of automatic chemical reactions within the chemistry society. This study proposes Chemist-X, a transformative AI agent that automates the reaction condition recommendation (RCR) task in chemical synthesis with retrieval-augmented generation (RAG) technology. To emulate expert chemists' strategies when solving RCR tasks, Chemist-X utilizes advanced RAG schemes to interrogate online molecular databases and distill critical data from the latest literature database. Further, the agent leverages state-of-the-art computer-aided design (CAD) tools with a large language model (LLM) supervised programming interface. With the ability to utilize updated chemical knowledge and CAD tools, our agent significantly outperforms conventional synthesis AIs confined to the fixed knowledge within its training data. Chemist-X considerably reduces chemists' workload and allows them to focus on more fundamental and creative problems, thereby bringing closer computational techniques and chemical research and making a remarkable leap toward harnessing AI's full capabilities in scientific discovery.
Read more4/5/2024