Uni-Mol Docking V2: Towards Realistic and Accurate Binding Pose Prediction

0

Sign in to get full access
Overview
- The paper presents Uni-Mol Docking V2, a method for accurately predicting the binding poses of molecules to protein targets.
- The approach aims to improve upon previous docking methods by incorporating more realistic physical and chemical interactions between the ligand and receptor.
- The researchers evaluate their method on a diverse dataset of protein-ligand complexes and compare its performance to other state-of-the-art docking tools.
Plain English Explanation
Uni-Mol Docking V2 is a new computer program that can predict how a small molecule (called a ligand) will bind to a larger protein (called a receptor). This is an important problem in drug discovery, because understanding how a potential drug molecule binds to its target can help researchers design more effective and safer drugs.
Previous docking methods have had trouble accurately predicting binding poses, often producing results that don't match experimental data. The researchers behind Uni-Mol Docking V2 have tried to address this by developing a more realistic model of the physical and chemical interactions between the ligand and receptor.
Their approach takes into account factors like the shape and charge distribution of the molecules, as well as the flexibility of the receptor. By modeling these details more accurately, Uni-Mol Docking V2 can generate binding predictions that are closer to what is observed in real-world experiments.
The researchers tested their method on a diverse set of protein-ligand complexes and found that it outperformed other leading docking tools. This suggests that Uni-Mol Docking V2 could be a valuable tool for drug discovery, helping researchers identify promising drug candidates more efficiently.
Technical Explanation
The key innovation in Uni-Mol Docking V2 is the use of a more sophisticated energy function to evaluate potential binding poses. Previous work has shown that accurately modeling the receptor's flexibility and the specific interactions between the ligand and receptor pockets is critical for realistic binding pose prediction.
To address this, the authors of Uni-Mol Docking V2 developed a new scoring function that incorporates detailed information about the shape, charge distribution, and solvation properties of both the ligand and receptor. This allows the method to better capture the complex physical and chemical forces driving molecular recognition.
Additionally, the authors leveraged large-scale pre-training of docking conformations to initialize their docking algorithm, which helps it converge to realistic binding poses more efficiently.
In their experiments, Uni-Mol Docking V2 demonstrated improved prediction of ligand-protein binding affinities compared to other state-of-the-art docking tools. The authors attribute this to their more accurate modeling of the key interactions driving molecular recognition.
Critical Analysis
The authors acknowledge that Uni-Mol Docking V2 still has some limitations. For example, the method currently assumes a rigid receptor structure, which may not always be accurate. Incorporating more flexibility into the receptor model could further improve the realism of the binding pose predictions.
Additionally, the dataset used for evaluating the method, while diverse, may not capture the full breadth of protein-ligand interactions encountered in real-world drug discovery scenarios. Expanding the testing to include a wider range of target classes and binding modes could help validate the broader applicability of Uni-Mol Docking V2.
That said, the authors' focus on developing a more physically and chemically grounded docking approach is a promising direction. Techniques like multimodal alignment of molecules and proteins and guiding docking along geodesic paths could further enhance the realism and accuracy of binding pose prediction, ultimately accelerating the drug discovery process.
Conclusion
Uni-Mol Docking V2 represents a significant advance in the field of molecular docking, demonstrating the potential for more physically and chemically accurate modeling to improve the realism of binding pose predictions. By better capturing the complex interplay of forces driving molecular recognition, the method shows promising results in outperforming other state-of-the-art docking tools.
While some limitations remain, the authors' focus on developing a more rigorous and grounded docking approach is an important step forward. As the field continues to evolve, techniques like Uni-Mol Docking V2 could play a crucial role in accelerating the drug discovery process and ultimately bringing more effective and safer medicines to patients.
This summary was produced with help from an AI and may contain inaccuracies - check out the links to read the original source documents!
Related Papers
0
Uni-Mol Docking V2: Towards Realistic and Accurate Binding Pose Prediction
Eric Alcaide, Zhifeng Gao, Guolin Ke, Yaqi Li, Linfeng Zhang, Hang Zheng, Gengmo Zhou
In recent years, machine learning (ML) methods have emerged as promising alternatives for molecular docking, offering the potential for high accuracy without incurring prohibitive computational costs. However, recent studies have indicated that these ML models may overfit to quantitative metrics while neglecting the physical constraints inherent in the problem. In this work, we present Uni-Mol Docking V2, which demonstrates a remarkable improvement in performance, accurately predicting the binding poses of 77+% of ligands in the PoseBusters benchmark with an RMSD value of less than 2.0 {AA}, and 75+% passing all quality checks. This represents a significant increase from the 62% achieved by the previous Uni-Mol Docking model. Notably, our Uni-Mol Docking approach generates chemically accurate predictions, circumventing issues such as chirality inversions and steric clashes that have plagued previous ML models. Furthermore, we observe enhanced performance in terms of high-quality predictions (RMSD values of less than 1.0 {AA} and 1.5 {AA}) and physical soundness when Uni-Mol Docking is combined with more physics-based methods like Uni-Dock. Our results represent a significant advancement in the application of artificial intelligence for scientific research, adopting a holistic approach to ligand docking that is well-suited for industrial applications in virtual screening and drug design. The code, data and service for Uni-Mol Docking are publicly available for use and further development in https://github.com/dptech-corp/Uni-Mol.
Read more5/21/2024

0
Smiles2Dock: an open large-scale multi-task dataset for ML-based molecular docking
Thomas Le Menestrel, Manuel Rivas
Docking is a crucial component in drug discovery aimed at predicting the binding conformation and affinity between small molecules and target proteins. ML-based docking has recently emerged as a prominent approach, outpacing traditional methods like DOCK and AutoDock Vina in handling the growing scale and complexity of molecular libraries. However, the availability of comprehensive and user-friendly datasets for training and benchmarking ML-based docking algorithms remains limited. We introduce Smiles2Dock, an open large-scale multi-task dataset for molecular docking. We created a framework combining P2Rank and AutoDock Vina to dock 1.7 million ligands from the ChEMBL database against 15 AlphaFold proteins, giving us more than 25 million protein-ligand binding scores. The dataset leverages a wide range of high-accuracy AlphaFold protein models, encompasses a diverse set of biologically relevant compounds and enables researchers to benchmark all major approaches for ML-based docking such as Graph, Transformer and CNN-based methods. We also introduce a novel Transformer-based architecture for docking scores prediction and set it as an initial benchmark for our dataset. Our dataset and code are publicly available to support the development of novel ML-based methods for molecular docking to advance scientific research in this field.
Read more6/11/2024
🤿
0
Deep Learning for Protein-Ligand Docking: Are We There Yet?
Alex Morehead, Nabin Giri, Jian Liu, Jianlin Cheng
The effects of ligand binding on protein structures and their in vivo functions carry numerous implications for modern biomedical research and biotechnology development efforts such as drug discovery. Although several deep learning (DL) methods and benchmarks designed for protein-ligand docking have recently been introduced, to date no prior works have systematically studied the behavior of docking methods within the practical context of (1) using predicted (apo) protein structures for docking (e.g., for broad applicability); (2) docking multiple ligands concurrently to a given target protein (e.g., for enzyme design); and (3) having no prior knowledge of binding pockets (e.g., for pocket generalization). To enable a deeper understanding of docking methods' real-world utility, we introduce PoseBench, the first comprehensive benchmark for practical protein-ligand docking. PoseBench enables researchers to rigorously and systematically evaluate DL docking methods for apo-to-holo protein-ligand docking and protein-ligand structure generation using both single and multi-ligand benchmark datasets, the latter of which we introduce for the first time to the DL community. Empirically, using PoseBench, we find that all recent DL docking methods but one fail to generalize to multi-ligand protein targets and also that template-based docking algorithms perform equally well or better for multi-ligand docking as recent single-ligand DL docking methods, suggesting areas of improvement for future work. Code, data, tutorials, and benchmark results are available at https://github.com/BioinfoMachineLearning/PoseBench.
Read more7/9/2024
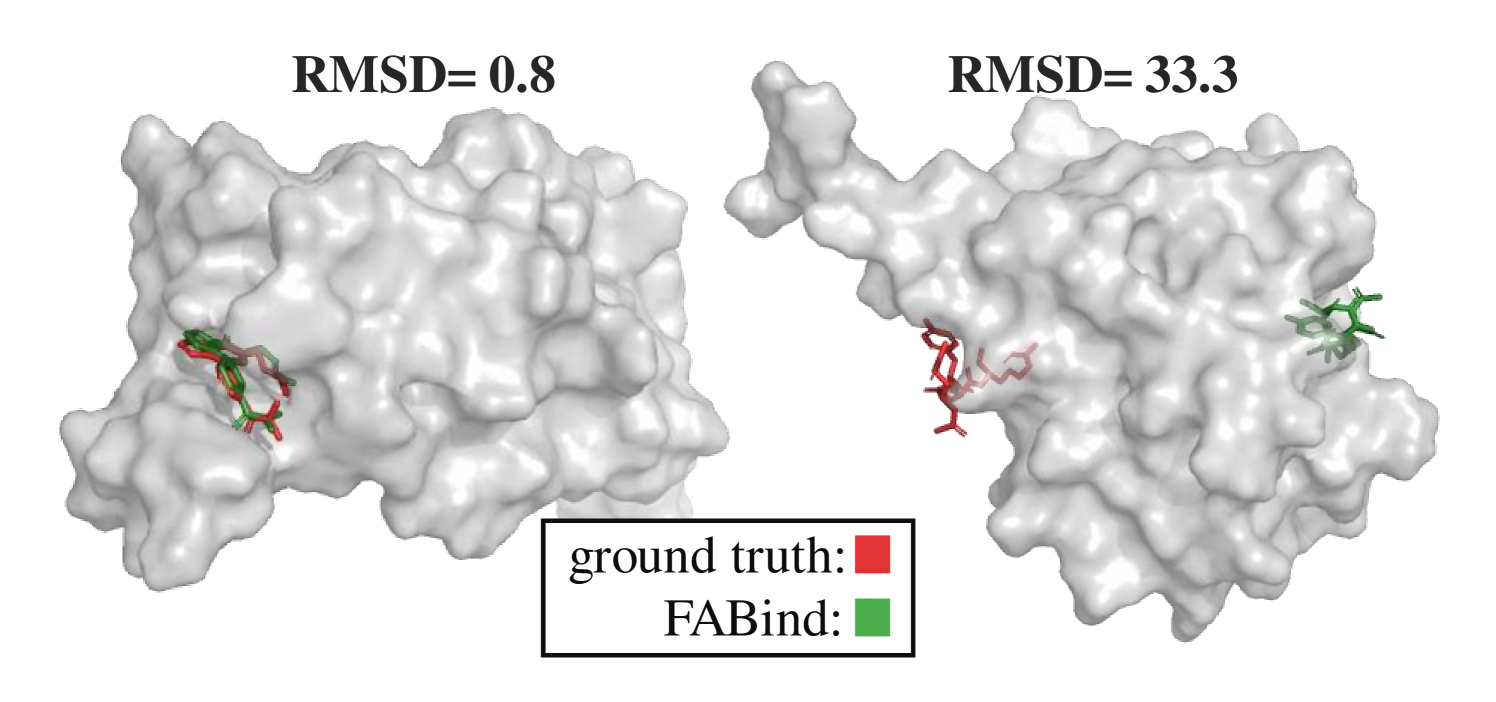
0
FABind+: Enhancing Molecular Docking through Improved Pocket Prediction and Pose Generation
Kaiyuan Gao, Qizhi Pei, Jinhua Zhu, Kun He, Lijun Wu
Molecular docking is a pivotal process in drug discovery. While traditional techniques rely on extensive sampling and simulation governed by physical principles, these methods are often slow and costly. The advent of deep learning-based approaches has shown significant promise, offering increases in both accuracy and efficiency. Building upon the foundational work of FABind, a model designed with a focus on speed and accuracy, we present FABind+, an enhanced iteration that largely boosts the performance of its predecessor. We identify pocket prediction as a critical bottleneck in molecular docking and propose a novel methodology that significantly refines pocket prediction, thereby streamlining the docking process. Furthermore, we introduce modifications to the docking module to enhance its pose generation capabilities. In an effort to bridge the gap with conventional sampling/generative methods, we incorporate a simple yet effective sampling technique coupled with a confidence model, requiring only minor adjustments to the regression framework of FABind. Experimental results and analysis reveal that FABind+ remarkably outperforms the original FABind, achieves competitive state-of-the-art performance, and delivers insightful modeling strategies. This demonstrates FABind+ represents a substantial step forward in molecular docking and drug discovery. Our code is in https://github.com/QizhiPei/FABind.
Read more4/9/2024