Accelerating the Training and Improving the Reliability of Machine-Learned Interatomic Potentials for Strongly Anharmonic Materials through Active Learning

0
Sign in to get full access
Overview
- Accelerating the training and improving the reliability of machine-learned interatomic potentials for strongly anharmonic materials through active learning
- Focuses on developing accurate and efficient interatomic potentials using machine learning techniques
- Addresses challenges in modeling strongly anharmonic materials, which are important for various applications
Plain English Explanation
Machine learning techniques have shown promise for developing accurate interatomic potentials, which are mathematical models that describe the interactions between atoms in a material. These models are crucial for simulating and understanding the behavior of materials at the atomic scale. However, constructing reliable interatomic potentials for strongly anharmonic materials, where atoms exhibit complex, non-linear vibrations, remains a significant challenge.
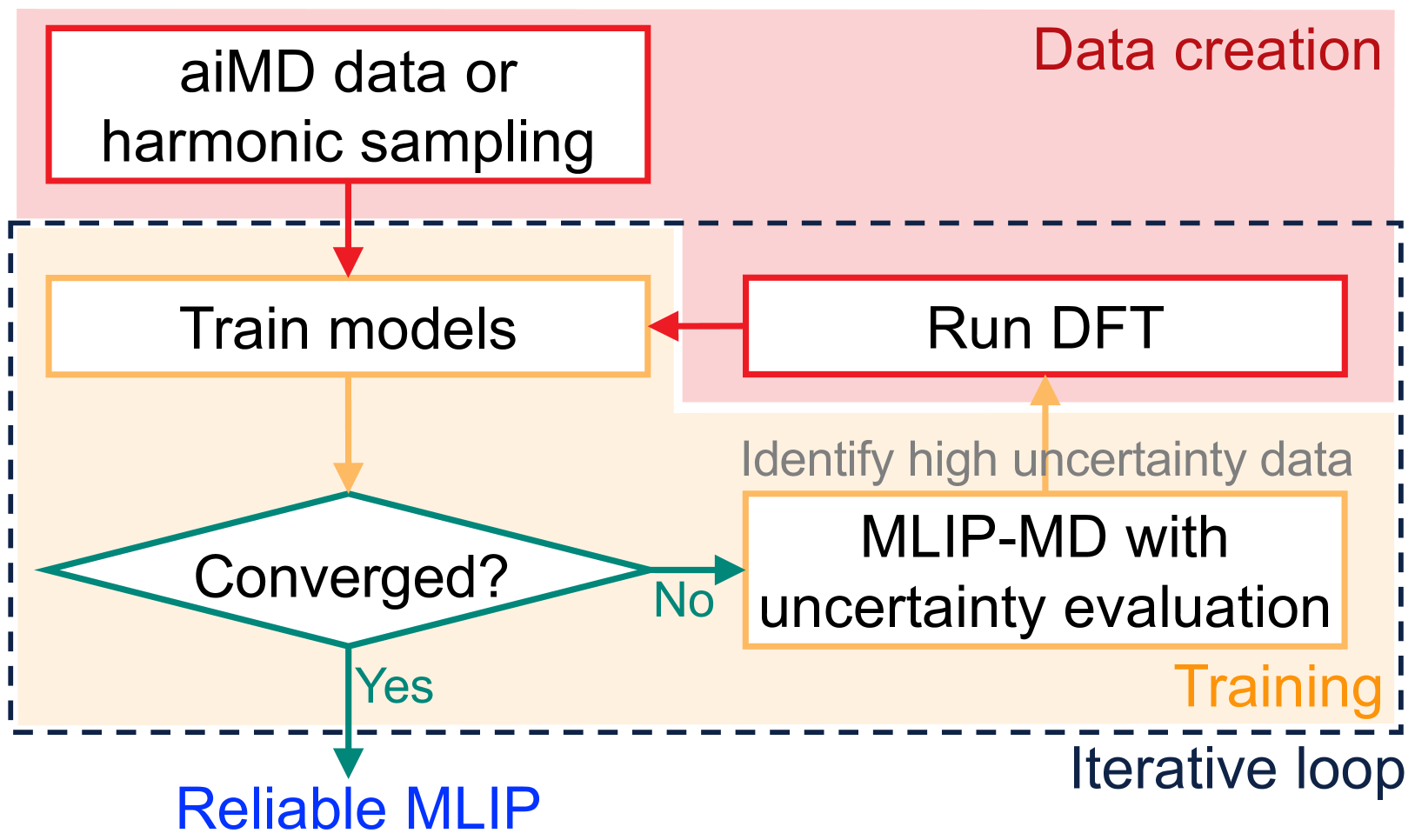
This research paper presents a novel approach to accelerate the training and improve the reliability of machine-learned interatomic potentials for strongly anharmonic materials. The key idea is to use an "active learning" strategy, where the model actively selects the most informative data points to include in the training process, rather than relying on a predefined dataset.
By focusing the training on the most crucial regions of the potential energy surface, the researchers were able to develop accurate interatomic potentials with fewer data points, reducing the computational cost and time required. This approach also helps to overcome the systematic softening often observed in machine-learned interatomic potentials, improving their reliability and predictive power.
The researchers demonstrated the effectiveness of their approach using several strongly anharmonic materials, such as high-entropy alloys and complex oxides, which are important for a wide range of applications, including energy storage, catalysis, and structural materials.
Technical Explanation
The researchers developed a physics-informed active learning framework to construct accurate and efficient machine-learned interatomic potentials for strongly anharmonic materials. The key components of their approach include:
-
Active Learning Strategy: The model actively selects the most informative data points to include in the training process, focusing on the regions of the potential energy surface that are crucial for accurately describing the material's behavior.
-
Physics-Informed Neural Networks: The researchers used neural network architectures that incorporate physical constraints and prior knowledge about the material, such as the symmetry of the atomic interactions.
-
Multifidelity Modeling: The approach combines data from different sources, including high-fidelity quantum-mechanical calculations and lower-fidelity classical simulations, to further improve the efficiency and accuracy of the interatomic potentials.
The researchers tested their approach on several strongly anharmonic materials, including high-entropy alloys and complex oxides, and demonstrated its ability to accelerate the training and improve the reliability of the resulting interatomic potentials, compared to traditional methods.
Critical Analysis
The researchers acknowledge that their approach relies on the availability of high-quality reference data, which can be computationally expensive to generate, especially for complex materials. Additionally, the active learning strategy may be sensitive to the initial selection of training data points, and further research is needed to understand the optimal sampling strategies for different types of materials.
While the paper demonstrates the effectiveness of the proposed approach on several strongly anharmonic materials, it would be interesting to see how it performs on an even broader range of materials, including those with different types of bonding and crystal structures. Additionally, the researchers could explore the integration of their approach with other techniques, such as transfer learning or multiscale modeling, to further enhance the efficiency and applicability of machine-learned interatomic potentials.
Conclusion
This research paper presents a novel approach to accelerate the training and improve the reliability of machine-learned interatomic potentials for strongly anharmonic materials. By incorporating physics-informed active learning strategies, the researchers were able to develop accurate and efficient interatomic potentials with fewer data points, overcoming the challenges associated with modeling complex, non-linear atomic interactions.
The implications of this work are significant, as accurate and efficient interatomic potentials are crucial for simulating and understanding the behavior of materials at the atomic scale, with applications in areas such as energy storage, catalysis, and structural materials. The proposed approach represents an important step forward in the field of computational materials science, and the insights gained from this research can inspire further advancements in the development of machine-learned interatomic potentials for a wide range of materials.
This summary was produced with help from an AI and may contain inaccuracies - check out the links to read the original source documents!
Related Papers
0
New!Accelerating the Training and Improving the Reliability of Machine-Learned Interatomic Potentials for Strongly Anharmonic Materials through Active Learning
Kisung Kang, Thomas A. R. Purcell, Christian Carbogno, Matthias Scheffler
Molecular dynamics (MD) employing machine-learned interatomic potentials (MLIPs) serve as an efficient, urgently needed complement to ab initio molecular dynamics (aiMD). By training these potentials on data generated from ab initio methods, their averaged predictions can exhibit comparable performance to ab initio methods at a fraction of the cost. However, insufficient training sets might lead to an improper description of the dynamics in strongly anharmonic materials, because critical effects might be overlooked in relevant cases, or only incorrectly captured, or hallucinated by the MLIP when they are not actually present. In this work, we show that an active learning scheme that combines MD with MLIPs (MLIP-MD) and uncertainty estimates can avoid such problematic predictions. In short, efficient MLIP-MD is used to explore configuration space quickly, whereby an acquisition function based on uncertainty estimates and on energetic viability is employed to maximize the value of the newly generated data and to focus on the most unfamiliar but reasonably accessible regions of phase space. To verify our methodology, we screen over 112 materials and identify 10 examples experiencing the aforementioned problems. Using CuI and AgGaSe$_2$ as archetypes for these problematic materials, we discuss the physical implications for strongly anharmonic effects and demonstrate how the developed active learning scheme can address these issues.
Read more9/19/2024

0
Physics-Informed Weakly Supervised Learning for Interatomic Potentials
Makoto Takamoto, Viktor Zaverkin, Mathias Niepert
Machine learning plays an increasingly important role in computational chemistry and materials science, complementing computationally intensive ab initio and first-principles methods. Despite their utility, machine-learning models often lack generalization capability and robustness during atomistic simulations, yielding unphysical energy and force predictions that hinder their real-world applications. We address this challenge by introducing a physics-informed, weakly supervised approach for training machine-learned interatomic potentials (MLIPs). We introduce two novel loss functions, extrapolating the potential energy via a Taylor expansion and using the concept of conservative forces. Our approach improves the accuracy of MLIPs applied to training tasks with sparse training data sets and reduces the need for pre-training computationally demanding models with large data sets. Particularly, we perform extensive experiments demonstrating reduced energy and force errors -- often lower by a factor of two -- for various baseline models and benchmark data sets. Finally, we show that our approach facilitates MLIPs' training in a setting where the computation of forces is infeasible at the reference level, such as those employing complete-basis-set extrapolation.
Read more8/13/2024
📊
0
Physics-informed active learning for accelerating quantum chemical simulations
Yi-Fan Hou, Lina Zhang, Quanhao Zhang, Fuchun Ge, Pavlo O. Dral
Quantum chemical simulations can be greatly accelerated by constructing machine learning potentials, which is often done using active learning (AL). The usefulness of the constructed potentials is often limited by the high effort required and their insufficient robustness in the simulations. Here we introduce the end-to-end AL for constructing robust data-efficient potentials with affordable investment of time and resources and minimum human interference. Our AL protocol is based on the physics-informed sampling of training points, automatic selection of initial data, uncertainty quantification, and convergence monitoring. The versatility of this protocol is shown in our implementation of quasi-classical molecular dynamics for simulating vibrational spectra, conformer search of a key biochemical molecule, and time-resolved mechanism of the Diels-Alder reactions. These investigations took us days instead of weeks of pure quantum chemical calculations on a high-performance computing cluster. The code in MLatom and tutorials are available at https://github.com/dralgroup/mlatom.
Read more7/17/2024

0
Overcoming systematic softening in universal machine learning interatomic potentials by fine-tuning
Bowen Deng, Yunyeong Choi, Peichen Zhong, Janosh Riebesell, Shashwat Anand, Zhuohan Li, KyuJung Jun, Kristin A. Persson, Gerbrand Ceder
Machine learning interatomic potentials (MLIPs) have introduced a new paradigm for atomic simulations. Recent advancements have seen the emergence of universal MLIPs (uMLIPs) that are pre-trained on diverse materials datasets, providing opportunities for both ready-to-use universal force fields and robust foundations for downstream machine learning refinements. However, their performance in extrapolating to out-of-distribution complex atomic environments remains unclear. In this study, we highlight a consistent potential energy surface (PES) softening effect in three uMLIPs: M3GNet, CHGNet, and MACE-MP-0, which is characterized by energy and force under-prediction in a series of atomic-modeling benchmarks including surfaces, defects, solid-solution energetics, phonon vibration modes, ion migration barriers, and general high-energy states. We find that the PES softening behavior originates from a systematic underprediction error of the PES curvature, which derives from the biased sampling of near-equilibrium atomic arrangements in uMLIP pre-training datasets. We demonstrate that the PES softening issue can be effectively rectified by fine-tuning with a single additional data point. Our findings suggest that a considerable fraction of uMLIP errors are highly systematic, and can therefore be efficiently corrected. This result rationalizes the data-efficient fine-tuning performance boost commonly observed with foundational MLIPs. We argue for the importance of a comprehensive materials dataset with improved PES sampling for next-generation foundational MLIPs.
Read more5/14/2024