Interpolation and differentiation of alchemical degrees of freedom in machine learning interatomic potentials
2404.10746
0
0
Abstract
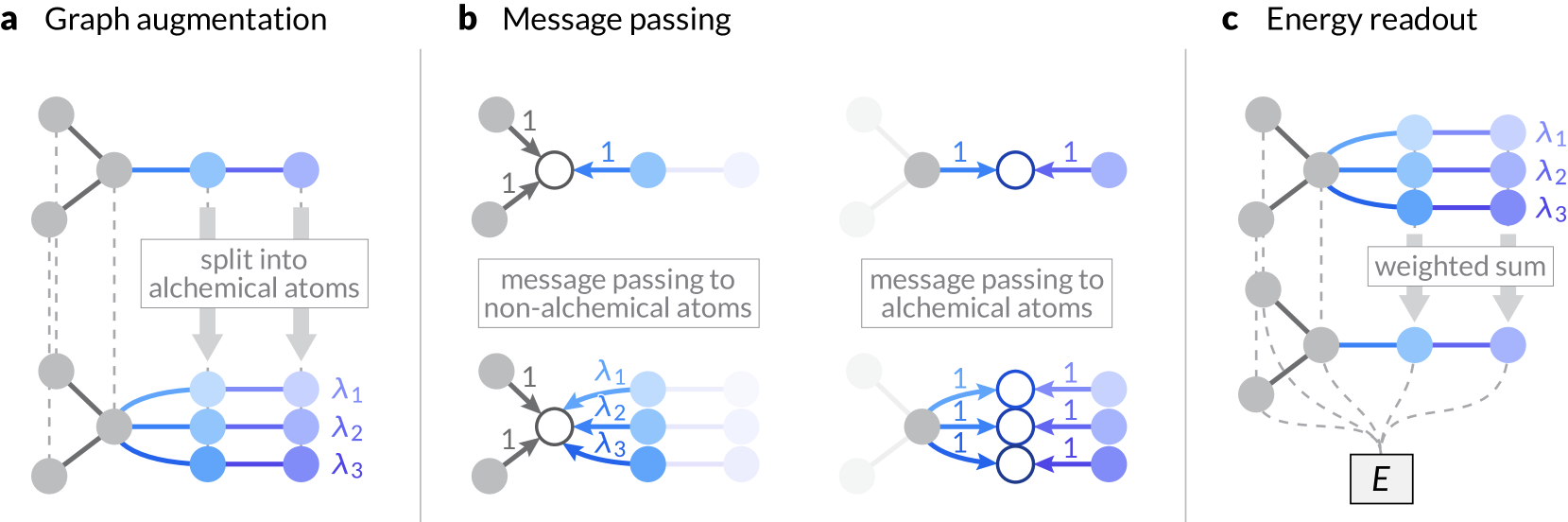
Machine learning interatomic potentials (MLIPs) have become a workhorse of modern atomistic simulations, and recently published universal MLIPs, pre-trained on large datasets, have demonstrated remarkable accuracy and generalizability. However, the computational cost of MLIPs limits their applicability to chemically disordered systems requiring large simulation cells or to sample-intensive statistical methods. Here, we report the use of continuous and differentiable alchemical degrees of freedom in atomistic materials simulations, exploiting the fact that graph neural network MLIPs represent discrete elements as real-valued tensors. The proposed method introduces alchemical atoms with corresponding weights into the input graph, alongside modifications to the message-passing and readout mechanisms of MLIPs, and allows smooth interpolation between the compositional states of materials. The end-to-end differentiability of MLIPs enables efficient calculation of the gradient of energy with respect to the compositional weights. Leveraging these gradients, we propose methodologies for optimizing the composition of solid solutions towards target macroscopic properties and conducting alchemical free energy simulations to quantify the free energy of vacancy formation and composition changes. The approach offers an avenue for extending the capabilities of universal MLIPs in the modeling of compositional disorder and characterizing the phase stabilities of complex materials systems.
Create account to get full access
Overview
- Addresses the challenge of interpolating and differentiating alchemical degrees of freedom in machine learning interatomic potentials
- Proposes a method to efficiently represent and manipulate these alchemical degrees of freedom
- Demonstrates how this approach can improve the accuracy and efficiency of molecular simulations
Plain English Explanation
Machine learning models are increasingly being used to simulate the interactions between atoms and molecules, known as interatomic potentials. These models can be very accurate, but they often have a large number of parameters that need to be tuned. Some of these parameters are "alchemical" degrees of freedom, which control how the model behaves when the chemical composition of the system is changed.
The provided paper presents a new method for efficiently representing and manipulating these alchemical degrees of freedom. The key idea is to use a "lightweight" representation that allows the model to be easily interpolated and differentiated with respect to the alchemical parameters. This means the model can adapt smoothly to changes in the chemical composition, which is important for tasks like catalyst design or materials discovery.
The authors demonstrate that their approach can improve the accuracy and efficiency of molecular simulations compared to previous methods. By making it easier to explore different chemical compositions, this work could help accelerate the development of new materials and catalysts with desirable properties.
Technical Explanation
The paper introduces a novel method for representing and manipulating the alchemical degrees of freedom in machine learning interatomic potentials. Alchemical degrees of freedom are parameters that control how a model's predictions change when the chemical composition of the system is varied.
The authors propose a "lightweight" representation of these alchemical degrees of freedom, which allows for efficient interpolation and differentiation. This is achieved by constructing a low-dimensional latent space that captures the relevant variations in the alchemical parameters. The model can then be conditioned on this latent representation, enabling smooth adaptation to changes in chemical composition.
The authors show that this approach outperforms standard methods for incorporating alchemical degrees of freedom, both in terms of accuracy and computational efficiency. The lightweight representation allows the model to be easily optimized and differentiated with respect to the alchemical parameters, which is crucial for applications like catalyst design and materials discovery.
The paper also demonstrates how this method can be integrated into a larger machine learning pipeline for molecular modeling, further enhancing its practical utility.
Critical Analysis
The paper presents a well-designed and thorough investigation of the proposed method for handling alchemical degrees of freedom in machine learning interatomic potentials. The authors have carefully considered the relevant prior work and have made a convincing case for the benefits of their approach.
One potential limitation is the reliance on a low-dimensional latent space to represent the alchemical degrees of freedom. While this strategy seems to work well in the examples provided, it may not be suitable for all types of chemical systems, especially those with more complex alchemical dependencies.
Additionally, the paper does not address the challenge of accurately estimating the uncertainty in the model's predictions when extrapolating to new alchemical regimes. This is an important consideration for applications where reliability and safety are paramount, such as in the design of new materials and catalysts.
Overall, the paper presents a promising approach that could significantly improve the efficiency and accuracy of molecular simulations involving changes in chemical composition. Further research and validation on a broader range of systems would help to strengthen the generalizability of the method.
Conclusion
This paper introduces a novel method for efficiently representing and manipulating the alchemical degrees of freedom in machine learning interatomic potentials. By using a lightweight latent space representation, the authors demonstrate improvements in both accuracy and computational efficiency compared to standard approaches.
The proposed technique has the potential to accelerate the discovery and development of new materials and catalysts, as it allows for the smooth exploration of different chemical compositions. This could be particularly impactful in fields like renewable energy, where the design of novel catalysts is crucial for improving the efficiency and sustainability of chemical processes.
Overall, this work represents an important step forward in the integration of machine learning with molecular modeling, and the authors have provided a solid foundation for future research and applications in this domain.
This summary was produced with help from an AI and may contain inaccuracies - check out the links to read the original source documents!
Related Papers

Overcoming systematic softening in universal machine learning interatomic potentials by fine-tuning
Bowen Deng, Yunyeong Choi, Peichen Zhong, Janosh Riebesell, Shashwat Anand, Zhuohan Li, KyuJung Jun, Kristin A. Persson, Gerbrand Ceder
0
0
Machine learning interatomic potentials (MLIPs) have introduced a new paradigm for atomic simulations. Recent advancements have seen the emergence of universal MLIPs (uMLIPs) that are pre-trained on diverse materials datasets, providing opportunities for both ready-to-use universal force fields and robust foundations for downstream machine learning refinements. However, their performance in extrapolating to out-of-distribution complex atomic environments remains unclear. In this study, we highlight a consistent potential energy surface (PES) softening effect in three uMLIPs: M3GNet, CHGNet, and MACE-MP-0, which is characterized by energy and force under-prediction in a series of atomic-modeling benchmarks including surfaces, defects, solid-solution energetics, phonon vibration modes, ion migration barriers, and general high-energy states. We find that the PES softening behavior originates from a systematic underprediction error of the PES curvature, which derives from the biased sampling of near-equilibrium atomic arrangements in uMLIP pre-training datasets. We demonstrate that the PES softening issue can be effectively rectified by fine-tuning with a single additional data point. Our findings suggest that a considerable fraction of uMLIP errors are highly systematic, and can therefore be efficiently corrected. This result rationalizes the data-efficient fine-tuning performance boost commonly observed with foundational MLIPs. We argue for the importance of a comprehensive materials dataset with improved PES sampling for next-generation foundational MLIPs.
5/14/2024

Neural Thermodynamic Integration: Free Energies from Energy-based Diffusion Models
B'alint M'at'e, Franc{c}ois Fleuret, Tristan Bereau
0
0
Thermodynamic integration (TI) offers a rigorous method for estimating free-energy differences by integrating over a sequence of interpolating conformational ensembles. However, TI calculations are computationally expensive and typically limited to coupling a small number of degrees of freedom due to the need to sample numerous intermediate ensembles with sufficient conformational-space overlap. In this work, we propose to perform TI along an alchemical pathway represented by a trainable neural network, which we term Neural TI. Critically, we parametrize a time-dependent Hamiltonian interpolating between the interacting and non-interacting systems, and optimize its gradient using a denoising-diffusion objective. The ability of the resulting energy-based diffusion model to sample all intermediate ensembles allows us to perform TI from a single reference calculation. We apply our method to Lennard-Jones fluids, where we report accurate calculations of the excess chemical potential, demonstrating that Neural TI is capable of coupling hundreds of degrees of freedom at once.
6/13/2024
↗️
Optimal design of experiments in the context of machine-learning inter-atomic potentials: improving the efficiency and transferability of kernel based methods
Bartosz Barzdajn, Christopher P. Race
0
0
Data-driven, machine learning (ML) models of atomistic interactions are often based on flexible and non-physical functions that can relate nuanced aspects of atomic arrangements into predictions of energies and forces. As a result, these potentials are as good as the training data (usually results of so-called ab initio simulations) and we need to make sure that we have enough information for a model to become sufficiently accurate, reliable and transferable. The main challenge stems from the fact that descriptors of chemical environments are often sparse high-dimensional objects without a well-defined continuous metric. Therefore, it is rather unlikely that any ad hoc method of choosing training examples will be indiscriminate, and it will be easy to fall into the trap of confirmation bias, where the same narrow and biased sampling is used to generate train- and test- sets. We will demonstrate that classical concepts of statistical planning of experiments and optimal design can help to mitigate such problems at a relatively low computational cost. The key feature of the method we will investigate is that they allow us to assess the informativeness of data (how much we can improve the model by adding/swapping a training example) and verify if the training is feasible with the current set before obtaining any reference energies and forces -- a so-called off-line approach. In other words, we are focusing on an approach that is easy to implement and doesn't require sophisticated frameworks that involve automated access to high-performance computational (HPC).
5/15/2024
📊
Physics-informed active learning for accelerating quantum chemical simulations
Yi-Fan Hou, Lina Zhang, Quanhao Zhang, Fuchun Ge, Pavlo O. Dral
0
0
Quantum chemical simulations can be greatly accelerated by constructing machine learning potentials, which is often done using active learning (AL). The usefulness of the constructed potentials is often limited by the high effort required and their insufficient robustness in the simulations. Here we introduce the end-to-end AL for constructing robust data-efficient potentials with affordable investment of time and resources and minimum human interference. Our AL protocol is based on the physics-informed sampling of training points, automatic selection of initial data, and uncertainty quantification. The versatility of this protocol is shown in our implementation of quasi-classical molecular dynamics for simulating vibrational spectra, conformer search of a key biochemical molecule, and time-resolved mechanism of the Diels-Alder reaction. These investigations took us days instead of weeks of pure quantum chemical calculations on a high-performance computing cluster.
4/19/2024