Bayesian optimization of atomic structures with prior probabilities from universal interatomic potentials

0
Sign in to get full access
Overview
- Bayesian optimization is used to find optimal atomic structures
- Prior probabilities from universal interatomic potentials are incorporated to guide the optimization
- This approach aims to efficiently explore the configuration space and identify promising candidate structures
Plain English Explanation
Designing new materials with specific atomic structures is an important challenge in materials science. Bayesian optimization is a powerful technique that can be used to search for the optimal atomic arrangement.
In this research, the scientists combine Bayesian optimization with prior information about interatomic interactions. They use universal interatomic potentials to estimate the likelihood of different atomic configurations before running expensive simulations or experiments. This helps guide the optimization process towards promising candidate structures more efficiently.
The key idea is to leverage our existing knowledge about how atoms interact, as captured in these universal potentials, to inform the search. This acts as a kind of "prior" that biases the optimization towards structures that are more physically plausible and likely to have desirable properties.
By incorporating this prior information, the researchers aim to explore the vast configuration space of possible atomic arrangements in a smarter, more targeted way. This could lead to the discovery of new materials with improved performance much faster than brute-force trial-and-error approaches.
Technical Explanation
The paper presents a Bayesian optimization framework that incorporates prior probabilities derived from universal interatomic potentials to guide the search for optimal atomic structures.
The key steps are:
- Define a Gaussian process model to represent the relationship between atomic configurations and their target properties.
- Use universal interatomic potentials to estimate the prior probability of different atomic arrangements.
- Combine the Gaussian process model with the prior probabilities to perform Bayesian optimization, iteratively updating the model and exploring promising regions of the configuration space.
- The algorithm efficiently identifies optimal or near-optimal atomic structures by leveraging both the Gaussian process surrogate model and the informative priors.
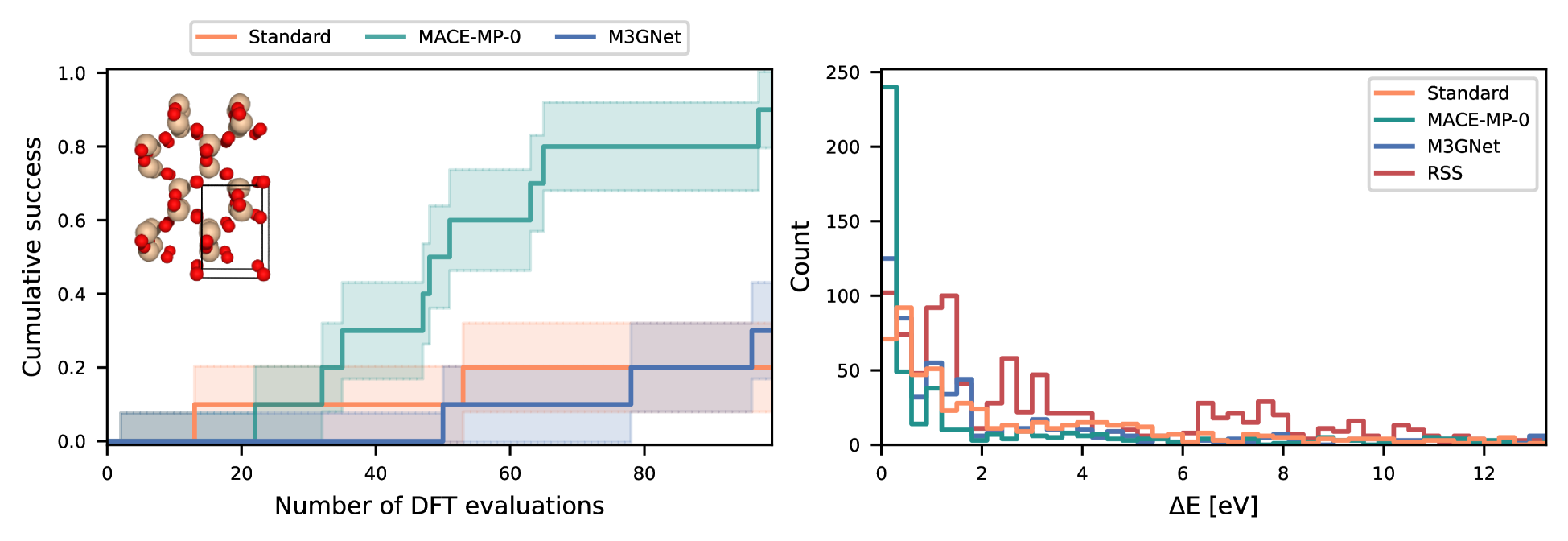
The researchers demonstrate the effectiveness of this approach on several materials design problems, showing that it can outperform traditional Bayesian optimization without the use of priors.
Critical Analysis
The proposed method relies heavily on the accuracy and generality of the universal interatomic potentials used to define the prior probabilities. While these potentials aim to capture universal trends in atomic interactions, their validity may be limited for certain classes of materials or extreme conditions.
Additionally, the Gaussian process model used in the Bayesian optimization framework may struggle to accurately represent highly complex, multi-modal objective functions that can arise in materials design problems. Further research is needed to explore the robustness of this approach to such challenges.
Finally, the computational cost of evaluating the universal potentials and integrating them into the Bayesian optimization loop could be non-trivial, especially for large-scale problems. The authors do not provide a detailed analysis of the scalability and runtime performance of their method.
Nevertheless, the core idea of leveraging prior information about interatomic interactions to guide materials design optimization is promising and could be further developed and refined in future work. Careful validation and comparison to other state-of-the-art techniques would be valuable to fully assess the merits and limitations of this approach.
Conclusion
This research presents a novel Bayesian optimization framework that incorporates prior probabilities from universal interatomic potentials to efficiently search for optimal atomic structures. By leveraging our existing knowledge about how atoms interact, the method aims to explore the configuration space in a more targeted and effective manner, potentially leading to faster discovery of new materials with desirable properties.
While the approach shows promise, further work is needed to address potential limitations and ensure its robustness across a wide range of materials design challenges. Continued advancements in this direction could have significant implications for accelerating materials innovation and supporting the development of novel, high-performance materials for various applications.
This summary was produced with help from an AI and may contain inaccuracies - check out the links to read the original source documents!
Related Papers
0
Bayesian optimization of atomic structures with prior probabilities from universal interatomic potentials
Peder Lyngby, Casper Larsen, Karsten Wedel Jacobsen
The optimization of atomic structures plays a pivotal role in understanding and designing materials with desired properties. However, conventional methods often struggle with the formidable task of navigating the vast potential energy surface, especially in high-dimensional spaces with numerous local minima. Recent advancements in machine learning-driven surrogate models offer a promising avenue for alleviating this computational burden. In this study, we propose a novel approach that combines the strengths of universal machine learning potentials with a Bayesian approach of the GOFEE/BEACON framework. By leveraging the comprehensive chemical knowledge encoded in pretrained universal machine learning potentials as a prior estimate of energy and forces, we enable the Gaussian process to focus solely on capturing the intricate nuances of the potential energy surface. We demonstrate the efficacy of our approach through comparative analyses across diverse systems, including periodic bulk materials, surface structures, and a cluster.
Read more8/29/2024
↗️
0
Optimal design of experiments in the context of machine-learning inter-atomic potentials: improving the efficiency and transferability of kernel based methods
Bartosz Barzdajn, Christopher P. Race
Data-driven, machine learning (ML) models of atomistic interactions are often based on flexible and non-physical functions that can relate nuanced aspects of atomic arrangements into predictions of energies and forces. As a result, these potentials are as good as the training data (usually results of so-called ab initio simulations) and we need to make sure that we have enough information for a model to become sufficiently accurate, reliable and transferable. The main challenge stems from the fact that descriptors of chemical environments are often sparse high-dimensional objects without a well-defined continuous metric. Therefore, it is rather unlikely that any ad hoc method of choosing training examples will be indiscriminate, and it will be easy to fall into the trap of confirmation bias, where the same narrow and biased sampling is used to generate train- and test- sets. We will demonstrate that classical concepts of statistical planning of experiments and optimal design can help to mitigate such problems at a relatively low computational cost. The key feature of the method we will investigate is that they allow us to assess the informativeness of data (how much we can improve the model by adding/swapping a training example) and verify if the training is feasible with the current set before obtaining any reference energies and forces -- a so-called off-line approach. In other words, we are focusing on an approach that is easy to implement and doesn't require sophisticated frameworks that involve automated access to high-performance computational (HPC).
Read more5/15/2024

0
Physics-Informed Weakly Supervised Learning for Interatomic Potentials
Makoto Takamoto, Viktor Zaverkin, Mathias Niepert
Machine learning plays an increasingly important role in computational chemistry and materials science, complementing computationally intensive ab initio and first-principles methods. Despite their utility, machine-learning models often lack generalization capability and robustness during atomistic simulations, yielding unphysical energy and force predictions that hinder their real-world applications. We address this challenge by introducing a physics-informed, weakly supervised approach for training machine-learned interatomic potentials (MLIPs). We introduce two novel loss functions, extrapolating the potential energy via a Taylor expansion and using the concept of conservative forces. Our approach improves the accuracy of MLIPs applied to training tasks with sparse training data sets and reduces the need for pre-training computationally demanding models with large data sets. Particularly, we perform extensive experiments demonstrating reduced energy and force errors -- often lower by a factor of two -- for various baseline models and benchmark data sets. Finally, we show that our approach facilitates MLIPs' training in a setting where the computation of forces is infeasible at the reference level, such as those employing complete-basis-set extrapolation.
Read more8/13/2024
🔍
0
Cartesian atomic cluster expansion for machine learning interatomic potentials
Bingqing Cheng
Machine learning interatomic potentials are revolutionizing large-scale, accurate atomistic modelling in material science and chemistry. Many potentials use atomic cluster expansion or equivariant message passing frameworks. Such frameworks typically use spherical harmonics as angular basis functions, and then use Clebsch-Gordan contraction to maintain rotational symmetry, which may introduce redundancies in representations and computational overhead. We propose an alternative: a Cartesian-coordinates-based atomic density expansion. This approach provides a complete set of polynormially indepedent features of atomic environments while maintaining interaction body orders. Additionally, we integrate low-dimensional embeddings of various chemical elements and inter-atomic message passing. The resulting potential, named Cartesian Atomic Cluster Expansion (CACE), exhibits good accuracy, stability, and generalizability. We validate its performance in diverse systems, including bulk water, small molecules, and 25-element high-entropy alloys.
Read more7/31/2024