Optimal design of experiments in the context of machine-learning inter-atomic potentials: improving the efficiency and transferability of kernel based methods
2405.08636
0
0
↗️
Abstract
Data-driven, machine learning (ML) models of atomistic interactions are often based on flexible and non-physical functions that can relate nuanced aspects of atomic arrangements into predictions of energies and forces. As a result, these potentials are as good as the training data (usually results of so-called ab initio simulations) and we need to make sure that we have enough information for a model to become sufficiently accurate, reliable and transferable. The main challenge stems from the fact that descriptors of chemical environments are often sparse high-dimensional objects without a well-defined continuous metric. Therefore, it is rather unlikely that any ad hoc method of choosing training examples will be indiscriminate, and it will be easy to fall into the trap of confirmation bias, where the same narrow and biased sampling is used to generate train- and test- sets. We will demonstrate that classical concepts of statistical planning of experiments and optimal design can help to mitigate such problems at a relatively low computational cost. The key feature of the method we will investigate is that they allow us to assess the informativeness of data (how much we can improve the model by adding/swapping a training example) and verify if the training is feasible with the current set before obtaining any reference energies and forces -- a so-called off-line approach. In other words, we are focusing on an approach that is easy to implement and doesn't require sophisticated frameworks that involve automated access to high-performance computational (HPC).
Create account to get full access
Overview
- Machine learning models of atomic interactions often use flexible, non-physical functions to capture the nuances of atomic arrangements and predict energies and forces.
- These models are only as good as the training data, usually from ab initio simulations, so it's crucial to ensure the data is sufficiently accurate, reliable, and transferable.
- The main challenge is that descriptors of chemical environments are often sparse, high-dimensional objects without a well-defined continuous metric, making it difficult to choose training examples indiscriminately and avoid confirmation bias.
- The paper proposes using classical concepts of statistical experiment planning and optimal design to mitigate these issues at a low computational cost.
Plain English Explanation
Machine learning models that predict the behavior of atoms and molecules often use flexible, non-physical functions to capture the complex relationships between the arrangement of atoms and the resulting energies and forces. These models are only as good as the data used to train them, which typically comes from ab initio simulations - sophisticated computer calculations of atomic and molecular properties.
The challenge is that the mathematical descriptions of the chemical environments around atoms are high-dimensional and lack a well-defined continuous metric, making it difficult to choose which examples to use for training the model. There's a risk of confirmation bias, where the same narrow and biased set of examples is used to both train and test the model.
To address this, the researchers suggest applying classical statistical techniques for planning experiments and designing optimal data collection. This allows them to assess how informative a potential training example would be, and verify if the training is feasible, before even obtaining the reference energies and forces. The key benefit is that this "offline" approach is easy to implement and doesn't require sophisticated high-performance computing frameworks.
Technical Explanation
The paper explores the use of statistical experiment design and optimal data selection to improve the training of machine learning (ML) models for predicting atomic interactions and properties. These data-driven ML models typically rely on flexible, non-physical functions that can capture nuanced aspects of atomic arrangements and translate them into predictions of energies and forces.
However, the accuracy, reliability, and transferability of these models are heavily dependent on the quality and breadth of the training data, which is often obtained from ab initio simulations. The main challenge stems from the fact that descriptors of chemical environments are sparse, high-dimensional objects without a well-defined continuous metric. This makes it difficult to choose training examples in an indiscriminate manner, leading to a risk of confirmation bias, where the same narrow and biased sampling is used for both training and testing.
To address this issue, the researchers propose leveraging classical concepts of statistical experiment planning and optimal design. The key feature of their approach is the ability to assess the informativeness of potential training data (i.e., how much the model can be improved by adding or swapping a training example) and verify the feasibility of the training process before obtaining any reference energies and forces. This "offline" approach allows for a relatively low computational cost compared to sophisticated frameworks that involve automated access to high-performance computing resources.
The researchers suggest that this methodology can help mitigate the challenges associated with sparse, high-dimensional descriptors of chemical environments and the risk of confirmation bias, which are common issues in the development of transferable machine learning models for molecular property predictions and [other applications in computational chemistry](https://aimodels.fyi/papers/arxiv/data-error-scaling-machine-learning-natural-discrete, https://aimodels.fyi/papers/arxiv/interpolation-differentiation-alchemical-degrees-freedom-machine-learning).
Critical Analysis
The researchers present a promising approach to addressing the challenges of training accurate and reliable machine learning models for atomic and molecular simulations. Their emphasis on leveraging classical statistical techniques for experiment planning and optimal data selection is a pragmatic solution that can be implemented without requiring sophisticated high-performance computing frameworks.
One potential limitation of the proposed approach is that it relies on the ability to accurately assess the informativeness of potential training data before obtaining the reference energies and forces. While this "offline" assessment can save computational resources, the accuracy of these informativeness estimates may be influenced by the choice of descriptors and the underlying assumptions of the model. Further research may be needed to ensure the robustness of this assessment process, especially for complex or diverse chemical systems.
Additionally, the paper does not provide a comprehensive evaluation of the proposed methodology across a wide range of applications or datasets. It would be valuable to see how the method performs in comparison to other data selection strategies, such as active learning or data error scaling, and whether the benefits hold true for different types of atomic interactions and molecular properties.
Overall, the researchers have presented a thoughtful and pragmatic approach to addressing a critical challenge in the development of accurate and reliable machine learning models for computational chemistry. Their work serves as a valuable contribution to the field and encourages further exploration of statistical experiment design techniques in the context of data-driven modeling of atomic and molecular phenomena.
Conclusion
This paper proposes a novel approach to training machine learning models for atomic and molecular simulations by leveraging classical statistical techniques for experiment planning and optimal data selection. The key advantage of this "offline" method is that it allows for the assessment of training data informativeness and feasibility before obtaining the reference energies and forces, thereby reducing the computational cost compared to more sophisticated frameworks.
The researchers highlight the challenges posed by the sparse, high-dimensional descriptors of chemical environments and the risk of confirmation bias in traditional data selection methods. Their proposed statistical approach aims to mitigate these issues and improve the accuracy, reliability, and transferability of the resulting machine learning models.
While the paper presents a promising solution, further research may be needed to fully evaluate the robustness and scalability of the method across a diverse range of applications in computational chemistry. Nevertheless, the researchers' work serves as an important contribution to the ongoing efforts to develop effective and efficient data-driven modeling techniques for the study of atomic and molecular phenomena.
This summary was produced with help from an AI and may contain inaccuracies - check out the links to read the original source documents!
Related Papers
📊
Physics-informed active learning for accelerating quantum chemical simulations
Yi-Fan Hou, Lina Zhang, Quanhao Zhang, Fuchun Ge, Pavlo O. Dral
0
0
Quantum chemical simulations can be greatly accelerated by constructing machine learning potentials, which is often done using active learning (AL). The usefulness of the constructed potentials is often limited by the high effort required and their insufficient robustness in the simulations. Here we introduce the end-to-end AL for constructing robust data-efficient potentials with affordable investment of time and resources and minimum human interference. Our AL protocol is based on the physics-informed sampling of training points, automatic selection of initial data, and uncertainty quantification. The versatility of this protocol is shown in our implementation of quasi-classical molecular dynamics for simulating vibrational spectra, conformer search of a key biochemical molecule, and time-resolved mechanism of the Diels-Alder reaction. These investigations took us days instead of weeks of pure quantum chemical calculations on a high-performance computing cluster.
4/19/2024
🏷️
Bayesian Adaptive Calibration and Optimal Design
Rafael Oliveira, Dino Sejdinovic, David Howard, Edwin Bonilla
0
0
The process of calibrating computer models of natural phenomena is essential for applications in the physical sciences, where plenty of domain knowledge can be embedded into simulations and then calibrated against real observations. Current machine learning approaches, however, mostly rely on rerunning simulations over a fixed set of designs available in the observed data, potentially neglecting informative correlations across the design space and requiring a large amount of simulations. Instead, we consider the calibration process from the perspective of Bayesian adaptive experimental design and propose a data-efficient algorithm to run maximally informative simulations within a batch-sequential process. At each round, the algorithm jointly estimates the parameters of the posterior distribution and optimal designs by maximising a variational lower bound of the expected information gain. The simulator is modelled as a sample from a Gaussian process, which allows us to correlate simulations and observed data with the unknown calibration parameters. We show the benefits of our method when compared to related approaches across synthetic and real-data problems.
5/24/2024
Transfer Learning for Molecular Property Predictions from Small Data Sets
Thorren Kirschbaum, Annika Bande
0
0
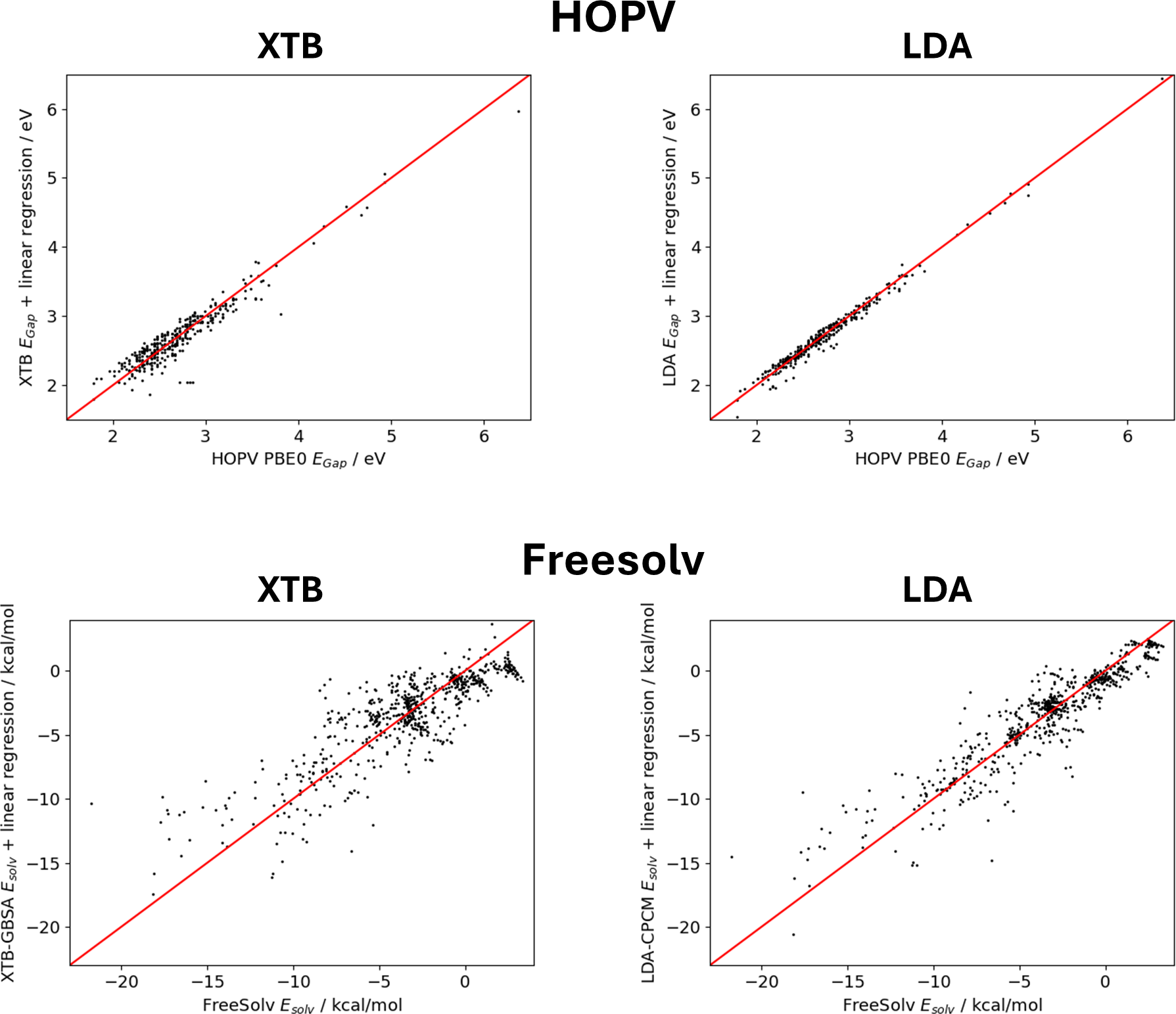
Machine learning has emerged as a new tool in chemistry to bypass expensive experiments or quantum-chemical calculations, for example, in high-throughput screening applications. However, many machine learning studies rely on small data sets, making it difficult to efficiently implement powerful deep learning architectures such as message passing neural networks. In this study, we benchmark common machine learning models for the prediction of molecular properties on small data sets, for which the best results are obtained with the message passing neural network PaiNN, as well as SOAP molecular descriptors concatenated to a set of simple molecular descriptors tailored to gradient boosting with regression trees. To further improve the predictive capabilities of PaiNN, we present a transfer learning strategy that uses large data sets to pre-train the respective models and allows to obtain more accurate models after fine-tuning on the original data sets. The pre-training labels are obtained from computationally cheap ab initio or semi-empirical models and corrected by simple linear regression on the target data set to obtain labels that are close to those of the original data. This strategy is tested on the Harvard Oxford Photovoltaics data set (HOPV, HOMO-LUMO-gaps), for which excellent results are obtained, and on the Freesolv data set (solvation energies), where this method is unsuccessful due to a complex underlying learning task and the dissimilar methods used to obtain pre-training and fine-tuning labels. Finally, we find that the final training results do not improve monotonically with the size of the pre-training data set, but pre-training with fewer data points can lead to more biased pre-trained models and higher accuracy after fine-tuning.
4/23/2024

Deep Optimal Experimental Design for Parameter Estimation Problems
Md Shahriar Rahim Siddiqui, Arman Rahmim, Eldad Haber
0
0
Optimal experimental design is a well studied field in applied science and engineering. Techniques for estimating such a design are commonly used within the framework of parameter estimation. Nonetheless, in recent years parameter estimation techniques are changing rapidly with the introduction of deep learning techniques to replace traditional estimation methods. This in turn requires the adaptation of optimal experimental design that is associated with these new techniques. In this paper we investigate a new experimental design methodology that uses deep learning. We show that the training of a network as a Likelihood Free Estimator can be used to significantly simplify the design process and circumvent the need for the computationally expensive bi-level optimization problem that is inherent in optimal experimental design for non-linear systems. Furthermore, deep design improves the quality of the recovery process for parameter estimation problems. As proof of concept we apply our methodology to two different systems of Ordinary Differential Equations.
6/26/2024