Enhancing material property prediction with ensemble deep graph convolutional networks

0

Sign in to get full access
Overview
- This paper explores the use of ensemble deep graph convolutional networks (EDGCNs) to enhance material property prediction.
- The key idea is to combine multiple deep learning models that analyze the structure of materials to make more accurate predictions of their properties.
- The approach is evaluated on several materials datasets, demonstrating improved performance compared to individual models.
Plain English Explanation
Materials such as metals, ceramics, and polymers have various properties like strength, conductivity, and reactivity that are important for engineering applications. Predicting these properties based on a material's atomic structure can be challenging, as the relationships between structure and properties can be complex.
The researchers in this paper propose using an ensemble of deep learning models, specifically deep graph convolutional networks, to tackle this challenge. These models are able to capture the intricate connections between the atoms in a material by representing the material as a graph.
By combining the predictions of multiple such models, the ensemble approach can make more accurate and robust predictions of material properties compared to using a single model. This is similar to how a group of experts can make better decisions than any one individual.
The researchers evaluate their ensemble approach on several real-world materials datasets, demonstrating clear performance improvements over individual models. This suggests that ensemble deep graph convolutional networks could be a powerful tool for accelerating the design and discovery of new materials with desirable properties.
Technical Explanation
The key technical contribution of this paper is the development of an ensemble deep graph convolutional network (EDGCN) for material property prediction. The EDGCN combines multiple graph neural network models, each of which learns to extract relevant features from the atomic structure of a material represented as a graph.
The ensemble approach works by training several base graph neural network models independently, each with a different architecture or training initialization. The predictions of these individual models are then combined, for example by averaging, to produce the final material property prediction.
The researchers evaluate their EDGCN approach on several materials datasets covering properties like band gap, formation energy, and atomization energy. They show that the ensemble model consistently outperforms individual graph neural network models, achieving state-of-the-art results.
The performance gains are attributed to the ensemble's ability to capture diverse representations of the materials and make more robust predictions by leveraging the complementary strengths of the base models. This echoes findings in other domains that ensemble methods can lead to better generalization.
Critical Analysis
The researchers provide a thorough evaluation of their ensemble approach, testing it on multiple materials datasets and comparing to strong individual baselines. The consistent performance improvements are compelling and suggest that EDGCN is a promising direction for enhancing material property prediction.
That said, the paper does not explore the limits of the approach. For example, it would be interesting to see how the ensemble performs as the number of base models is varied, or how it compares to other ensemble techniques like bagging or boosting. The researchers also do not delve into the interpretability of the ensemble model and how the individual base models contribute to the final predictions.
Additionally, the paper focuses on traditional material properties like band gap and formation energy. It would be valuable to extend the approach to other types of materials, such as those relevant for energy or biomedical applications, to further demonstrate its versatility and real-world impact.
Conclusion
This paper presents a novel ensemble deep learning approach for enhancing material property prediction. By combining multiple graph neural network models, the EDGCN framework can make more accurate and robust predictions of important material characteristics.
The strong empirical results on several datasets suggest that ensemble methods could be a powerful tool for accelerating materials discovery and design. As the field of computational materials science continues to evolve, techniques like EDGCN that leverage the complementary strengths of diverse models will likely play an increasingly important role.
This summary was produced with help from an AI and may contain inaccuracies - check out the links to read the original source documents!
Related Papers
0
Enhancing material property prediction with ensemble deep graph convolutional networks
Chowdhury Mohammad Abid Rahman, Ghadendra Bhandari, Nasser M Nasrabadi, Aldo H. Romero, Prashnna K. Gyawali
Machine learning (ML) models have emerged as powerful tools for accelerating materials discovery and design by enabling accurate predictions of properties from compositional and structural data. These capabilities are vital for developing advanced technologies across fields such as energy, electronics, and biomedicine, potentially reducing the time and resources needed for new material exploration and promoting rapid innovation cycles. Recent efforts have focused on employing advanced ML algorithms, including deep learning - based graph neural network, for property prediction. Additionally, ensemble models have proven to enhance the generalizability and robustness of ML and DL. However, the use of such ensemble strategies in deep graph networks for material property prediction remains underexplored. Our research provides an in-depth evaluation of ensemble strategies in deep learning - based graph neural network, specifically targeting material property prediction tasks. By testing the Crystal Graph Convolutional Neural Network (CGCNN) and its multitask version, MT-CGCNN, we demonstrated that ensemble techniques, especially prediction averaging, substantially improve precision beyond traditional metrics for key properties like formation energy per atom ($Delta E^{f}$), band gap ($E_{g}$) and density ($rho$) in 33,990 stable inorganic materials. These findings support the broader application of ensemble methods to enhance predictive accuracy in the field.
Read more7/29/2024
🧠
0
Hybrid Quantum Graph Neural Network for Molecular Property Prediction
Michael Vitz, Hamed Mohammadbagherpoor, Samarth Sandeep, Andrew Vlasic, Richard Padbury, Anh Pham
To accelerate the process of materials design, materials science has increasingly used data driven techniques to extract information from collected data. Specially, machine learning (ML) algorithms, which span the ML discipline, have demonstrated ability to predict various properties of materials with the level of accuracy similar to explicit calculation of quantum mechanical theories, but with significantly reduced run time and computational resources. Within ML, graph neural networks have emerged as an important algorithm within the field of machine learning, since they are capable of predicting accurately a wide range of important physical, chemical and electronic properties due to their higher learning ability based on the graph representation of material and molecular descriptors through the aggregation of information embedded within the graph. In parallel with the development of state of the art classical machine learning applications, the fusion of quantum computing and machine learning have created a new paradigm where classical machine learning model can be augmented with quantum layers which are able to encode high dimensional data more efficiently. Leveraging the structure of existing algorithms, we developed a unique and novel gradient free hybrid quantum classical convoluted graph neural network (HyQCGNN) to predict formation energies of perovskite materials. The performance of our hybrid statistical model is competitive with the results obtained purely from a classical convoluted graph neural network, and other classical machine learning algorithms, such as XGBoost. Consequently, our study suggests a new pathway to explore how quantum feature encoding and parametric quantum circuits can yield drastic improvements of complex ML algorithm like graph neural network.
Read more5/9/2024

0
Graph Residual based Method for Molecular Property Prediction
Kanad Sen, Saksham Gupta, Abhishek Raj, Alankar Alankar
Property prediction of materials has recently been of high interest in the recent years in the field of material science. Various Physics-based and Machine Learning models have already been developed, that can give good results. However, they are not accurate enough and are inadequate for critical applications. The traditional machine learning models try to predict properties based on the features extracted from the molecules, which are not easily available most of the time. In this paper, a recently developed novel Deep Learning method, the Graph Neural Network (GNN), has been applied, allowing us to predict properties directly only the Graph-based structures of the molecules. SMILES (Simplified Molecular Input Line Entry System) representation of the molecules has been used in the present study as input data format, which has been further converted into a graph database, which constitutes the training data. This article highlights the detailed description of the novel GRU-based methodology to map the inputs that have been used. Emphasis on highlighting both the regressive property as well as the classification-based property of the GNN backbone. A detailed description of the Variational Autoencoder (VAE) and the end-to-end learning method has been given to highlight the multi-class multi-label property prediction of the backbone. The results have been compared with standard benchmark datasets as well as some newly developed datasets. All performance metrics which have been used have been clearly defined as well as their reason for choice. Keywords: GNN, VAE, SMILES, multi-label multi-class classification, GRU
Read more8/9/2024
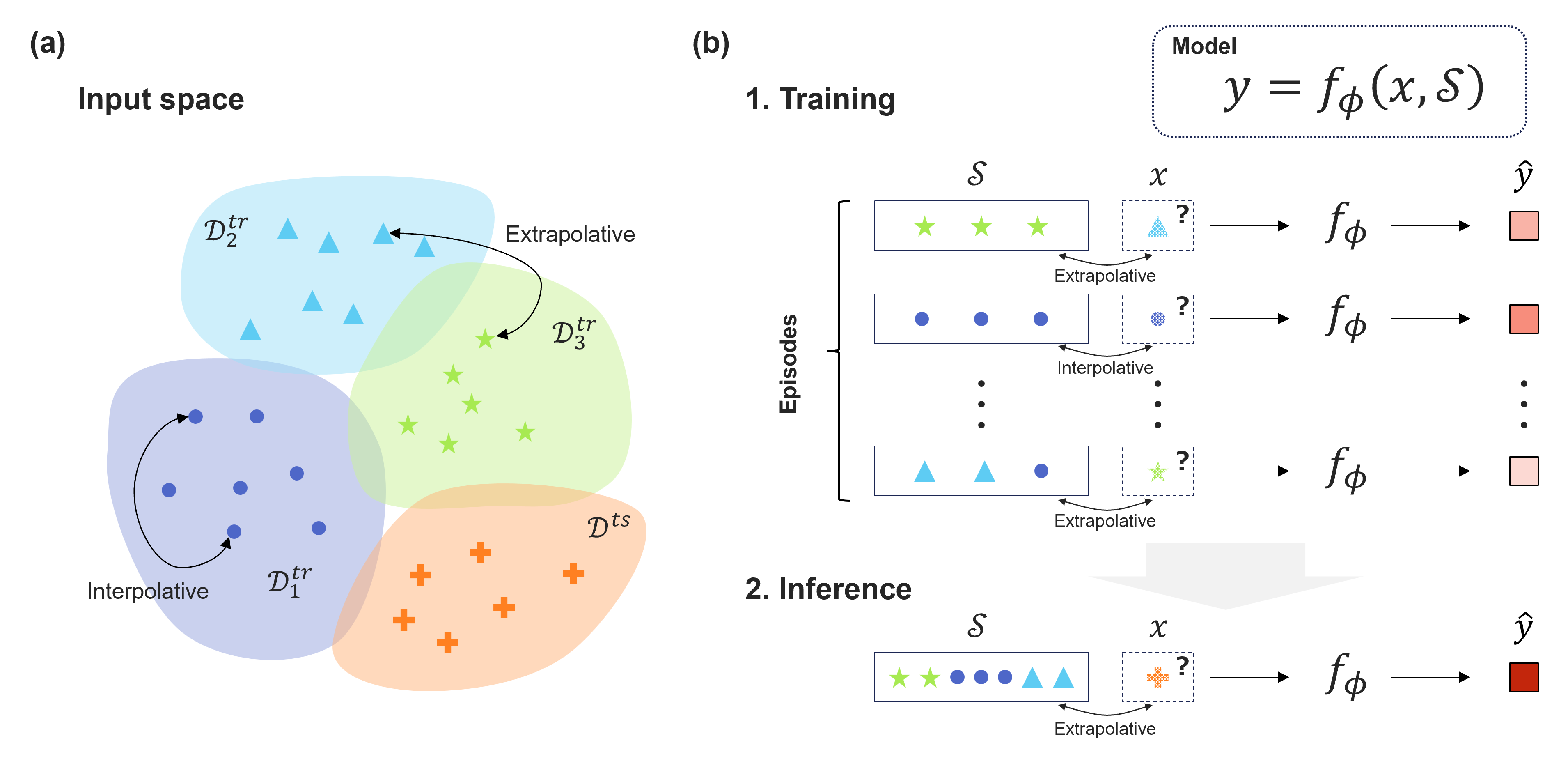
0
Advancing Extrapolative Predictions of Material Properties through Learning to Learn
Kohei Noda, Araki Wakiuchi, Yoshihiro Hayashi, Ryo Yoshida
Recent advancements in machine learning have showcased its potential to significantly accelerate the discovery of new materials. Central to this progress is the development of rapidly computable property predictors, enabling the identification of novel materials with desired properties from vast material spaces. However, the limited availability of data resources poses a significant challenge in data-driven materials research, particularly hindering the exploration of innovative materials beyond the boundaries of existing data. While machine learning predictors are inherently interpolative, establishing a general methodology to create an extrapolative predictor remains a fundamental challenge, limiting the search for innovative materials beyond existing data boundaries. In this study, we leverage an attention-based architecture of neural networks and meta-learning algorithms to acquire extrapolative generalization capability. The meta-learners, experienced repeatedly with arbitrarily generated extrapolative tasks, can acquire outstanding generalization capability in unexplored material spaces. Through the tasks of predicting the physical properties of polymeric materials and hybrid organic--inorganic perovskites, we highlight the potential of such extrapolatively trained models, particularly with their ability to rapidly adapt to unseen material domains in transfer learning scenarios.
Read more4/16/2024