Hierarchical novel class discovery for single-cell transcriptomic profiles

0

Sign in to get full access
Overview
- Introduces a novel hierarchical approach for discovering cell types from single-cell transcriptomic data
- Automatically discovers novel cell classes without relying on predefined cell type labels
- Outperforms existing methods on various real-world datasets
Plain English Explanation
The paper presents a new method for discovering novel cell types from single-cell RNA sequencing data. This is an important problem because identifying distinct cell populations is crucial for understanding the complex cellular composition of biological tissues.
Traditionally, researchers have relied on predefined cell type labels to group cells into known categories. However, this approach can miss previously uncharacterized or novel cell types. The proposed method aims to address this limitation by automatically discovering novel cell classes without requiring any prior information about cell types.
The key idea is to organize the cells in a hierarchical manner, allowing the algorithm to identify both broad cell lineages and more granular subpopulations. This hierarchical structure provides a more nuanced understanding of the cellular landscape compared to flat clustering approaches.
The method was evaluated on several real-world single-cell datasets and was shown to outperform existing techniques in terms of discovering biologically meaningful cell types and aligning with expert-annotated cell labels.
Technical Explanation
The paper introduces a hierarchical novel class discovery (HNCD) framework for single-cell transcriptomic data analysis. The core of the approach is a novel clustering algorithm that can automatically discover a hierarchical organization of cell types without relying on any predefined cell labels.
The method works by first embedding the single-cell transcriptomic profiles into a low-dimensional latent space using a deep neural network. It then applies a divisive hierarchical clustering algorithm to organize the cells into a tree-like structure, where each node represents a distinct cell type or subpopulation.
The key innovation is the use of a multimodal analysis of the data, which combines information about both the cell-cell similarity and the gene expression patterns. This allows the algorithm to identify biologically meaningful cell types that may not be readily apparent from the gene expression data alone.
The performance of HNCD was extensively evaluated on several benchmark single-cell datasets, where it demonstrated superior cell type discovery capabilities compared to existing methods. The discovered cell types were also found to align well with expert-annotated labels, suggesting the biological relevance of the identified cell populations.
Critical Analysis
The paper presents a compelling approach for discovering novel cell types from single-cell transcriptomic data. The hierarchical clustering strategy is a key strength, as it provides a more nuanced understanding of the cellular landscape compared to flat clustering methods.
However, the paper does not extensively discuss the limitations or potential issues with the proposed approach. For example, the method's performance may be sensitive to the choice of hyperparameters or the quality of the initial cell embedding. Additionally, the computational complexity of the hierarchical clustering algorithm could be a concern for extremely large datasets.
Further research could explore ways to make the method more robust and scalable, such as by incorporating techniques from the field of kernel-based testing for single-cell differential analysis. Validating the biological relevance of the discovered cell types through additional experiments or expert validation would also strengthen the claims made in the paper.
Conclusion
The hierarchical novel class discovery framework presented in this paper represents a promising advance in the field of single-cell transcriptomic analysis. By automatically discovering novel cell types without relying on predefined labels, the method provides a more comprehensive and unbiased understanding of the cellular composition of biological tissues.
The strong performance of HNCD on various real-world datasets suggests that it could be a valuable tool for researchers studying cell differentiation, cell-cell interactions, and other complex biological phenomena at the single-cell level. Further refinement and validation of the method could lead to even more impactful applications in the field of computational biology and bioinformatics.
This summary was produced with help from an AI and may contain inaccuracies - check out the links to read the original source documents!
Related Papers
0
Hierarchical novel class discovery for single-cell transcriptomic profiles
Malek Senoussi, Thierry Arti`eres, Paul Villoutreix
One of the major challenges arising from single-cell transcriptomics experiments is the question of how to annotate the associated single-cell transcriptomic profiles. Because of the large size and the high dimensionality of the data, automated methods for annotation are needed. We focus here on datasets obtained in the context of developmental biology, where the differentiation process leads to a hierarchical structure. We consider a frequent setting where both labeled and unlabeled data are available at training time, but the sets of the labels of labeled data on one side and of the unlabeled data on the other side, are disjoint. It is an instance of the Novel Class Discovery problem. The goal is to achieve two objectives, clustering the data and mapping the clusters with labels. We propose extensions of k-Means and GMM clustering methods for solving the problem and report comparative results on artificial and experimental transcriptomic datasets. Our approaches take advantage of the hierarchical nature of the data.
Read more9/11/2024
0
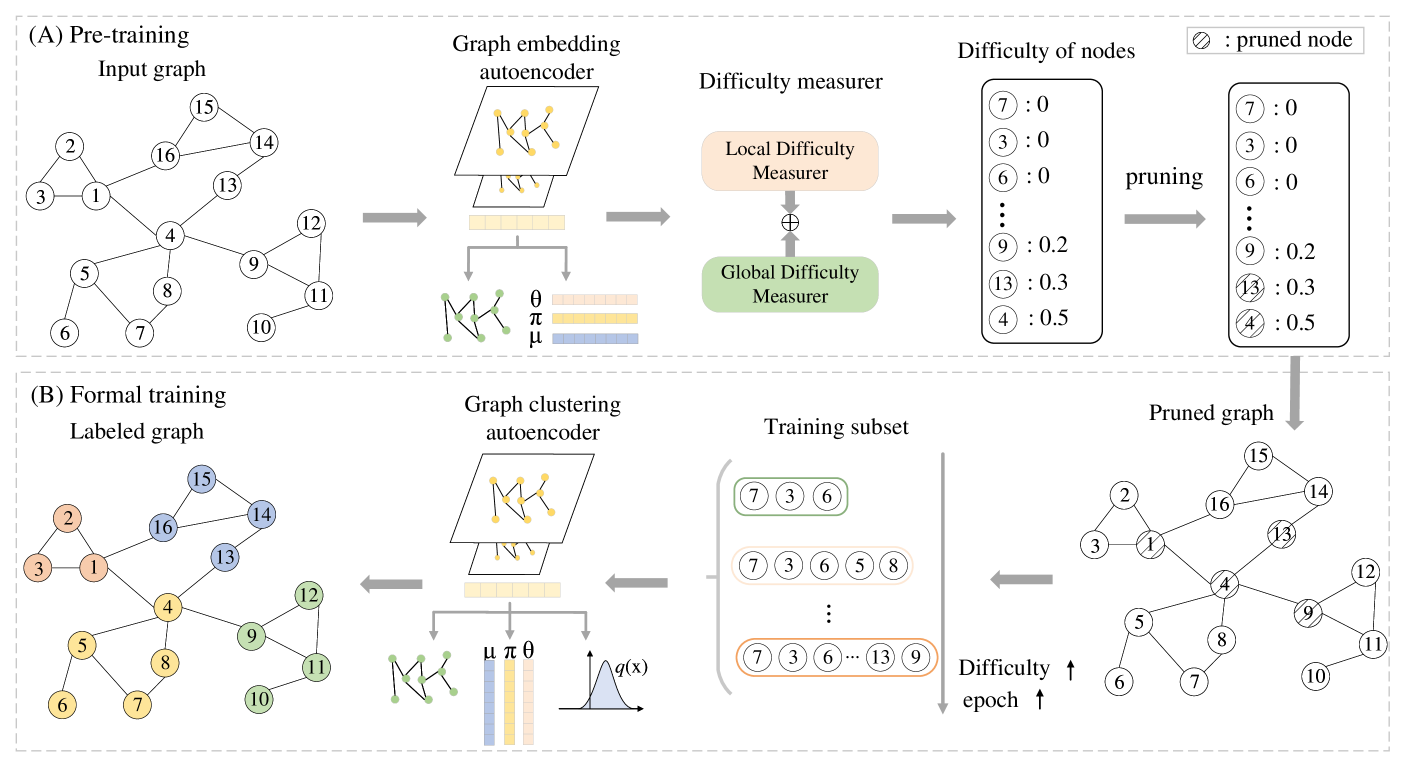
Single-cell Curriculum Learning-based Deep Graph Embedding Clustering
Huifa Li, Jie Fu, Xinpeng Ling, Zhiyu Sun, Kuncan Wang, Zhili Chen
The swift advancement of single-cell RNA sequencing (scRNA-seq) technologies enables the investigation of cellular-level tissue heterogeneity. Cell annotation significantly contributes to the extensive downstream analysis of scRNA-seq data. However, The analysis of scRNA-seq for biological inference presents challenges owing to its intricate and indeterminate data distribution, characterized by a substantial volume and a high frequency of dropout events. Furthermore, the quality of training samples varies greatly, and the performance of the popular scRNA-seq data clustering solution GNN could be harmed by two types of low-quality training nodes: 1) nodes on the boundary; 2) nodes that contribute little additional information to the graph. To address these problems, we propose a single-cell curriculum learning-based deep graph embedding clustering (scCLG). We first propose a Chebyshev graph convolutional autoencoder with multi-decoder (ChebAE) that combines three optimization objectives corresponding to three decoders, including topology reconstruction loss of cell graphs, zero-inflated negative binomial (ZINB) loss, and clustering loss, to learn cell-cell topology representation. Meanwhile, we employ a selective training strategy to train GNN based on the features and entropy of nodes and prune the difficult nodes based on the difficulty scores to keep the high-quality graph. Empirical results on a variety of gene expression datasets show that our model outperforms state-of-the-art methods.
Read more8/21/2024

0
scGHSOM: Hierarchical clustering and visualization of single-cell and CRISPR data using growing hierarchical SOM
Shang-Jung Wen, Jia-Ming Chang, Fang Yu
High-dimensional single-cell data poses significant challenges in identifying underlying biological patterns due to the complexity and heterogeneity of cellular states. We propose a comprehensive gene-cell dependency visualization via unsupervised clustering, Growing Hierarchical Self-Organizing Map (GHSOM), specifically designed for analyzing high-dimensional single-cell data like single-cell sequencing and CRISPR screens. GHSOM is applied to cluster samples in a hierarchical structure such that the self-growth structure of clusters satisfies the required variations between and within. We propose a novel Significant Attributes Identification Algorithm to identify features that distinguish clusters. This algorithm pinpoints attributes with minimal variation within a cluster but substantial variation between clusters. These key attributes can then be used for targeted data retrieval and downstream analysis. Furthermore, we present two innovative visualization tools: Cluster Feature Map and Cluster Distribution Map. The Cluster Feature Map highlights the distribution of specific features across the hierarchical structure of GHSOM clusters. This allows for rapid visual assessment of cluster uniqueness based on chosen features. The Cluster Distribution Map depicts leaf clusters as circles on the GHSOM grid, with circle size reflecting cluster data size and color customizable to visualize features like cell type or other attributes. We apply our analysis to three single-cell datasets and one CRISPR dataset (cell-gene database) and evaluate clustering methods with internal and external CH and ARI scores. GHSOM performs well, being the best performer in internal evaluation (CH=4.2). In external evaluation, GHSOM has the third-best performance of all methods.
Read more7/25/2024
🧪
0
Kernel-Based Testing for Single-Cell Differential Analysis
Anthony Ozier-Lafontaine, Camille Fourneaux, Ghislain Durif, Polina Arsenteva, C'eline Vallot, Olivier Gandrillon, Sandrine Giraud, Bertrand Michel, Franck Picard
Single-cell technologies offer insights into molecular feature distributions, but comparing them poses challenges. We propose a kernel-testing framework for non-linear cell-wise distribution comparison, analyzing gene expression and epigenomic modifications. Our method allows feature-wise and global transcriptome/epigenome comparisons, revealing cell population heterogeneities. Using a classifier based on embedding variability, we identify transitions in cell states, overcoming limitations of traditional single-cell analysis. Applied to single-cell ChIP-Seq data, our approach identifies untreated breast cancer cells with an epigenomic profile resembling persister cells. This demonstrates the effectiveness of kernel testing in uncovering subtle population variations that might be missed by other methods.
Read more4/15/2024