Single-cell Curriculum Learning-based Deep Graph Embedding Clustering

0
Sign in to get full access
Overview
- Single-cell RNA sequencing (scRNA-seq) data analysis is crucial for understanding cellular heterogeneity and identifying novel cell types.
- Graph-based clustering methods have shown promise for scRNA-seq data analysis, but can be sensitive to initialization and suffer from local optima.
- The paper proposes a Single-cell Curriculum Learning-based Deep Graph Embedding Clustering (SCDCG) method to address these issues.
Plain English Explanation
The paper introduces a new approach called SCDCG for analyzing single-cell RNA sequencing (scRNA-seq) data. scRNA-seq data provides a detailed snapshot of the gene activity within individual cells, allowing researchers to study the diversity and complexity of cell types in a tissue or organism.
One common way to analyze scRNA-seq data is by using graph-based clustering methods. These methods represent the cells as nodes in a graph, with edges connecting cells that are similar to each other. The goal is to then identify clusters of cells that are more connected to each other than to cells in other clusters, which often correspond to distinct cell types.
However, traditional graph-based clustering methods can have some limitations. They can be sensitive to the initial starting point, meaning the clusters they identify can vary depending on how the algorithm is initialized. They can also get stuck in local optima, where the clustering solution is good but not the best possible one.
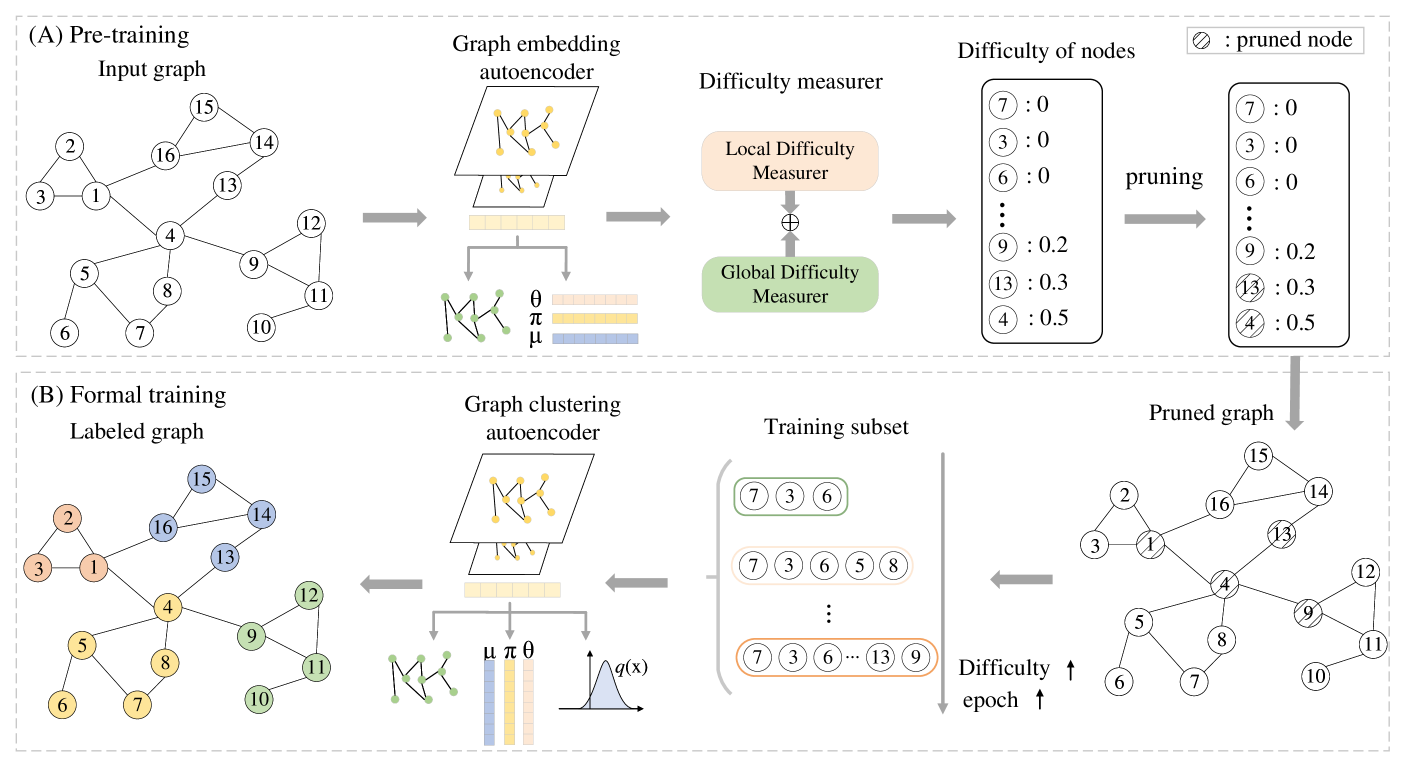
To address these issues, the SCDCG method uses a curriculum learning approach. Curriculum learning is a machine learning technique that trains a model in stages, starting with simpler tasks and gradually increasing the difficulty. In the case of SCDCG, the algorithm starts by clustering the cells using a simple, easy-to-optimize graph structure, and then progressively increases the complexity of the graph to obtain a more accurate clustering.
By using this curriculum learning strategy, SCDCG is able to avoid getting stuck in local optima and find a more globally optimal clustering solution for the scRNA-seq data. The authors demonstrate that SCDCG outperforms other state-of-the-art graph clustering and scRNA-seq analysis methods on a variety of benchmark datasets.
Technical Explanation
The SCDCG method consists of three main components:
-
Graph Embedding: The first step is to learn a low-dimensional representation, or embedding, of the cells in the scRNA-seq data. This is done using a deep neural network-based graph embedding model.
-
Curriculum Learning: The graph embedding model is trained using a curriculum learning strategy. The algorithm starts with a simple, easy-to-optimize graph structure and gradually increases the complexity of the graph over the course of training.
-
Clustering: The final step is to perform clustering on the learned cell embeddings. The authors use a spectral clustering approach to identify the final cell clusters, which correspond to distinct cell types.
The key insight behind the SCDCG method is that by using a curriculum learning approach, the graph embedding model can avoid getting stuck in local optima and instead converge to a more globally optimal solution. This leads to better clustering performance compared to other state-of-the-art methods.
The authors evaluate SCDCG on several scRNA-seq datasets and show that it outperforms other graph-based and deep learning-based clustering methods in terms of clustering accuracy, stability, and computational efficiency.
Critical Analysis
One potential limitation of the SCDCG method is that it relies on the assumption that the underlying cell population can be well-represented by a graph structure. If the true data manifold is not well-captured by a graph, the method may struggle to find the optimal clustering solution.
Additionally, the curriculum learning approach used in SCDCG requires careful design of the curriculum, which can be challenging and time-consuming. The authors do not provide detailed guidelines on how to set the curriculum parameters, which may make it difficult for other researchers to replicate their results.
It would also be valuable to see how SCDCG performs on larger and more complex scRNA-seq datasets, as the experiments in the paper were limited to relatively small-scale benchmarks. Scaling the method to handle datasets with millions of cells would be an important test of its practical applicability.
Conclusion
The SCDCG method presented in this paper is a promising approach for analyzing single-cell RNA sequencing data. By using a curriculum learning strategy to train a deep graph embedding model, the method is able to overcome some of the limitations of traditional graph-based clustering approaches and identify cell types more accurately.
The strong performance of SCDCG on benchmark datasets suggests that it could be a valuable tool for researchers studying cellular heterogeneity and uncovering novel cell types. Further research is needed to explore the method's scalability and robustness, but the core ideas behind SCDCG represent an important step forward in the field of single-cell data analysis.
This summary was produced with help from an AI and may contain inaccuracies - check out the links to read the original source documents!
Related Papers
0
Single-cell Curriculum Learning-based Deep Graph Embedding Clustering
Huifa Li, Jie Fu, Xinpeng Ling, Zhiyu Sun, Kuncan Wang, Zhili Chen
The swift advancement of single-cell RNA sequencing (scRNA-seq) technologies enables the investigation of cellular-level tissue heterogeneity. Cell annotation significantly contributes to the extensive downstream analysis of scRNA-seq data. However, The analysis of scRNA-seq for biological inference presents challenges owing to its intricate and indeterminate data distribution, characterized by a substantial volume and a high frequency of dropout events. Furthermore, the quality of training samples varies greatly, and the performance of the popular scRNA-seq data clustering solution GNN could be harmed by two types of low-quality training nodes: 1) nodes on the boundary; 2) nodes that contribute little additional information to the graph. To address these problems, we propose a single-cell curriculum learning-based deep graph embedding clustering (scCLG). We first propose a Chebyshev graph convolutional autoencoder with multi-decoder (ChebAE) that combines three optimization objectives corresponding to three decoders, including topology reconstruction loss of cell graphs, zero-inflated negative binomial (ZINB) loss, and clustering loss, to learn cell-cell topology representation. Meanwhile, we employ a selective training strategy to train GNN based on the features and entropy of nodes and prune the difficult nodes based on the difficulty scores to keep the high-quality graph. Empirical results on a variety of gene expression datasets show that our model outperforms state-of-the-art methods.
Read more8/21/2024

0
scCDCG: Efficient Deep Structural Clustering for single-cell RNA-seq via Deep Cut-informed Graph Embedding
Ping Xu, Zhiyuan Ning, Meng Xiao, Guihai Feng, Xin Li, Yuanchun Zhou, Pengfei Wang
Single-cell RNA sequencing (scRNA-seq) is essential for unraveling cellular heterogeneity and diversity, offering invaluable insights for bioinformatics advancements. Despite its potential, traditional clustering methods in scRNA-seq data analysis often neglect the structural information embedded in gene expression profiles, crucial for understanding cellular correlations and dependencies. Existing strategies, including graph neural networks, face challenges in handling the inefficiency due to scRNA-seq data's intrinsic high-dimension and high-sparsity. Addressing these limitations, we introduce scCDCG (single-cell RNA-seq Clustering via Deep Cut-informed Graph), a novel framework designed for efficient and accurate clustering of scRNA-seq data that simultaneously utilizes intercellular high-order structural information. scCDCG comprises three main components: (i) A graph embedding module utilizing deep cut-informed techniques, which effectively captures intercellular high-order structural information, overcoming the over-smoothing and inefficiency issues prevalent in prior graph neural network methods. (ii) A self-supervised learning module guided by optimal transport, tailored to accommodate the unique complexities of scRNA-seq data, specifically its high-dimension and high-sparsity. (iii) An autoencoder-based feature learning module that simplifies model complexity through effective dimension reduction and feature extraction. Our extensive experiments on 6 datasets demonstrate scCDCG's superior performance and efficiency compared to 7 established models, underscoring scCDCG's potential as a transformative tool in scRNA-seq data analysis. Our code is available at: https://github.com/XPgogogo/scCDCG.
Read more4/10/2024

0
scASDC: Attention Enhanced Structural Deep Clustering for Single-cell RNA-seq Data
Wenwen Min, Zhen Wang, Fangfang Zhu, Taosheng Xu, Shunfang Wang
Single-cell RNA sequencing (scRNA-seq) data analysis is pivotal for understanding cellular heterogeneity. However, the high sparsity and complex noise patterns inherent in scRNA-seq data present significant challenges for traditional clustering methods. To address these issues, we propose a deep clustering method, Attention-Enhanced Structural Deep Embedding Graph Clustering (scASDC), which integrates multiple advanced modules to improve clustering accuracy and robustness.Our approach employs a multi-layer graph convolutional network (GCN) to capture high-order structural relationships between cells, termed as the graph autoencoder module. To mitigate the oversmoothing issue in GCNs, we introduce a ZINB-based autoencoder module that extracts content information from the data and learns latent representations of gene expression. These modules are further integrated through an attention fusion mechanism, ensuring effective combination of gene expression and structural information at each layer of the GCN. Additionally, a self-supervised learning module is incorporated to enhance the robustness of the learned embeddings. Extensive experiments demonstrate that scASDC outperforms existing state-of-the-art methods, providing a robust and effective solution for single-cell clustering tasks. Our method paves the way for more accurate and meaningful analysis of single-cell RNA sequencing data, contributing to better understanding of cellular heterogeneity and biological processes. All code and public datasets used in this paper are available at url{https://github.com/wenwenmin/scASDC} and url{https://zenodo.org/records/12814320}.
Read more8/13/2024

0
Gene Regulatory Network Inference from Pre-trained Single-Cell Transcriptomics Transformer with Joint Graph Learning
Sindhura Kommu, Yizhi Wang, Yue Wang, Xuan Wang
Inferring gene regulatory networks (GRNs) from single-cell RNA sequencing (scRNA-seq) data is a complex challenge that requires capturing the intricate relationships between genes and their regulatory interactions. In this study, we tackle this challenge by leveraging the single-cell BERT-based pre-trained transformer model (scBERT), trained on extensive unlabeled scRNA-seq data, to augment structured biological knowledge from existing GRNs. We introduce a novel joint graph learning approach that combines the rich contextual representations learned by pre-trained single-cell language models with the structured knowledge encoded in GRNs using graph neural networks (GNNs). By integrating these two modalities, our approach effectively reasons over boththe gene expression level constraints provided by the scRNA-seq data and the structured biological knowledge inherent in GRNs. We evaluate our method on human cell benchmark datasets from the BEELINE study with cell type-specific ground truth networks. The results demonstrate superior performance over current state-of-the-art baselines, offering a deeper understanding of cellular regulatory mechanisms.
Read more7/26/2024