A high-accuracy multi-model mixing retrosynthetic method

0

Sign in to get full access
Overview
- This paper presents a novel single-step model for retrosynthetic reaction prediction.
- The model achieves state-of-the-art performance on several benchmark datasets.
- The key contributions include an improved encoder-decoder architecture and a new training approach.
Plain English Explanation
The paper describes a single-step model for retrosynthetic reaction prediction. Retrosynthetic analysis is the process of working backwards from a desired product to identify possible starting materials and synthetic steps. This model aims to simplify this complex task by predicting the full synthetic route in a single step, rather than requiring multiple sequential predictions.
The core innovation is an improved encoder-decoder architecture that can more effectively capture the relationships between reactants, products, and reaction conditions. The authors also introduce a new training approach that helps the model learn robust representations.
The results show that this single-step model outperforms previous state-of-the-art approaches on several standard benchmarks for retrosynthetic reaction prediction. This suggests it could be a valuable tool for organic chemists and pharmaceutical researchers working on complex synthetic challenges.
Technical Explanation
The paper presents a single-step model for retrosynthetic reaction prediction. The key technical components include:
-
Encoder-Decoder Architecture: The model uses a transformer-based encoder-decoder architecture to capture the complex relationships between reactants, products, and reaction conditions.
-
Training Approach: The authors introduce a new training approach that helps the model learn robust representations, enabling it to generalize better to unseen reactions.
-
Benchmark Evaluation: The model is evaluated on several standard datasets for retrosynthetic reaction prediction, including the USPTO and SYNTHIA benchmarks. The results show state-of-the-art performance compared to previous methods.
Critical Analysis
The paper makes a compelling case for the benefits of a single-step retrosynthetic prediction model. However, some potential limitations and areas for further research are worth considering:
- The model's performance may be sensitive to the quality and coverage of the training data. Expanding the diversity of the datasets could help improve generalization.
- The approach relies on transformer-based architectures, which can be computationally intensive. Exploring more efficient model designs could be valuable for real-world applications.
- The paper does not address the model's interpretability or its ability to provide chemists with insights into the underlying reaction mechanisms. Enhancing the model's explanatory capabilities could further increase its usefulness.
Overall, this research represents an important step forward in retrosynthetic reaction prediction and could have significant implications for streamlining the drug discovery and development process.
Conclusion
This paper presents a novel single-step model for retrosynthetic reaction prediction that achieves state-of-the-art performance on several benchmark datasets. The key technical innovations include an improved encoder-decoder architecture and a new training approach. While the model shows promise, potential limitations around data quality, computational efficiency, and interpretability could be addressed in future research to further enhance its practical utility for organic chemists and pharmaceutical researchers.
This summary was produced with help from an AI and may contain inaccuracies - check out the links to read the original source documents!
Related Papers
0
A high-accuracy multi-model mixing retrosynthetic method
Shang Xiang, Lin Yao, Zhen Wang, Qifan Yu, Wentan Liu, Wentao Guo, Guolin Ke
The field of computer-aided synthesis planning (CASP) has seen rapid advancements in recent years, achieving significant progress across various algorithmic benchmarks. However, chemists often encounter numerous infeasible reactions when using CASP in practice. This article delves into common errors associated with CASP and introduces a product prediction model aimed at enhancing the accuracy of single-step models. While the product prediction model reduces the number of single-step reactions, it integrates multiple single-step models to maintain the overall reaction count and increase reaction diversity. Based on manual analysis and large-scale testing, the product prediction model, combined with the multi-model ensemble approach, has been proven to offer higher feasibility and greater diversity.
Read more9/9/2024
🛸
0
DirectMultiStep: Direct Route Generation for Multi-Step Retrosynthesis
Yu Shee, Haote Li, Anton Morgunov, Victor Batista
Traditional computer-aided synthesis planning (CASP) methods rely on iterative single-step predictions, leading to exponential search space growth that limits efficiency and scalability. We introduce a transformer-based model that directly generates multi-step synthetic routes as a single string by conditionally predicting each molecule based on all preceding ones. The model accommodates specific conditions such as the desired number of steps and starting materials, outperforming state-of-the-art methods on the PaRoutes dataset with a 2.2x improvement in Top-1 accuracy on the n$_1$ test set and a 3.3x improvement on the n$_5$ test set. It also successfully predicts routes for FDA-approved drugs not included in the training data, showcasing its generalization capabilities. While the current suboptimal diversity of the training set may impact performance on less common reaction types, our approach presents a promising direction towards fully automated retrosynthetic planning.
Read more5/24/2024
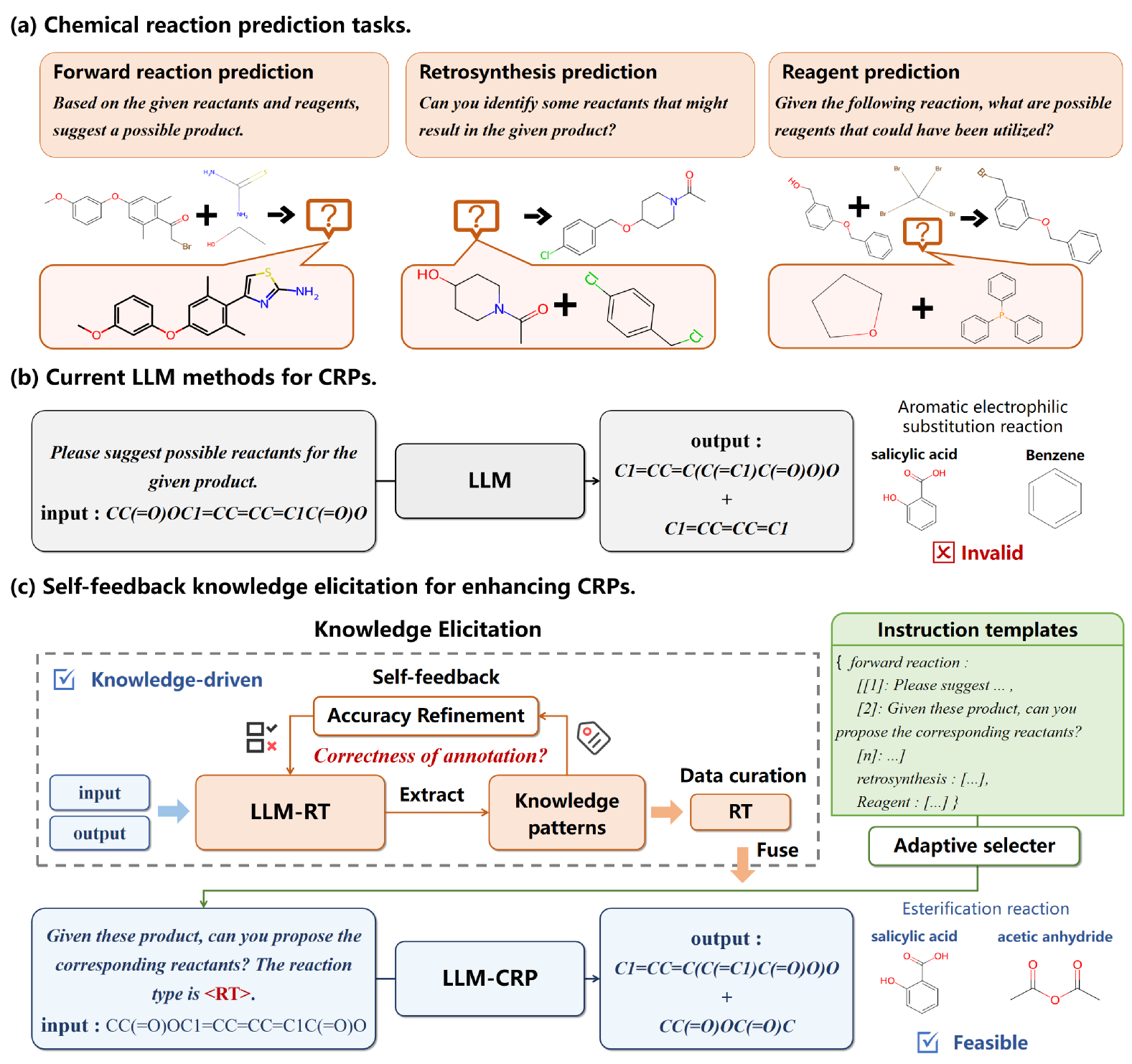
0
A Self-feedback Knowledge Elicitation Approach for Chemical Reaction Predictions
Pengfei Liu, Jun Tao, Zhixiang Ren
The task of chemical reaction predictions (CRPs) plays a pivotal role in advancing drug discovery and material science. However, its effectiveness is constrained by the vast and uncertain chemical reaction space and challenges in capturing reaction selectivity, particularly due to existing methods' limitations in exploiting the data's inherent knowledge. To address these challenges, we introduce a data-curated self-feedback knowledge elicitation approach. This method starts from iterative optimization of molecular representations and facilitates the extraction of knowledge on chemical reaction types (RTs). Then, we employ adaptive prompt learning to infuse the prior knowledge into the large language model (LLM). As a result, we achieve significant enhancements: a 14.2% increase in retrosynthesis prediction accuracy, a 74.2% rise in reagent prediction accuracy, and an expansion in the model's capability for handling multi-task chemical reactions. This research offers a novel paradigm for knowledge elicitation in scientific research and showcases the untapped potential of LLMs in CRPs.
Read more4/16/2024

0
Double-Ended Synthesis Planning with Goal-Constrained Bidirectional Search
Kevin Yu, Jihye Roh, Ziang Li, Wenhao Gao, Runzhong Wang, Connor W. Coley
Computer-aided synthesis planning (CASP) algorithms have demonstrated expert-level abilities in planning retrosynthetic routes to molecules of low to moderate complexity. However, current search methods assume the sufficiency of reaching arbitrary building blocks, failing to address the common real-world constraint where using specific molecules is desired. To this end, we present a formulation of synthesis planning with starting material constraints. Under this formulation, we propose Double-Ended Synthesis Planning (DESP), a novel CASP algorithm under a bidirectional graph search scheme that interleaves expansions from the target and from the goal starting materials to ensure constraint satisfiability. The search algorithm is guided by a goal-conditioned cost network learned offline from a partially observed hypergraph of valid chemical reactions. We demonstrate the utility of DESP in improving solve rates and reducing the number of search expansions by biasing synthesis planning towards expert goals on multiple new benchmarks. DESP can make use of existing one-step retrosynthesis models, and we anticipate its performance to scale as these one-step model capabilities improve.
Read more7/10/2024