Higher-Order Equivariant Neural Networks for Charge Density Prediction in Materials

0
Sign in to get full access
Overview
- This paper presents the development of higher-order equivariant neural networks for predicting the charge density of materials.
- The proposed approach aims to improve the accuracy and efficiency of charge density prediction, which is crucial for understanding and designing new materials.
- The researchers leverage the inherent symmetries of atomic structures to design neural network architectures that are equivariant to certain transformations, such as rotations and translations.
Plain English Explanation
The charge density of a material is a fundamental property that describes how electrons are distributed within the material. Accurately predicting charge density is important for understanding the properties and behavior of materials, which is essential for developing new materials with desirable characteristics.
This paper introduces a new approach to predicting charge density using machine learning. The researchers developed a type of neural network called "higher-order equivariant neural networks" that can take into account the symmetries of atomic structures. This means the neural network can recognize when the material is rotated or translated, and adjust its predictions accordingly.
By incorporating these symmetries, the researchers were able to create a more accurate and efficient model for predicting charge density. This could lead to better understanding of materials and accelerate the development of new materials with improved properties, such as those used in molecular modeling and catalyst design or metamaterial design.
Technical Explanation
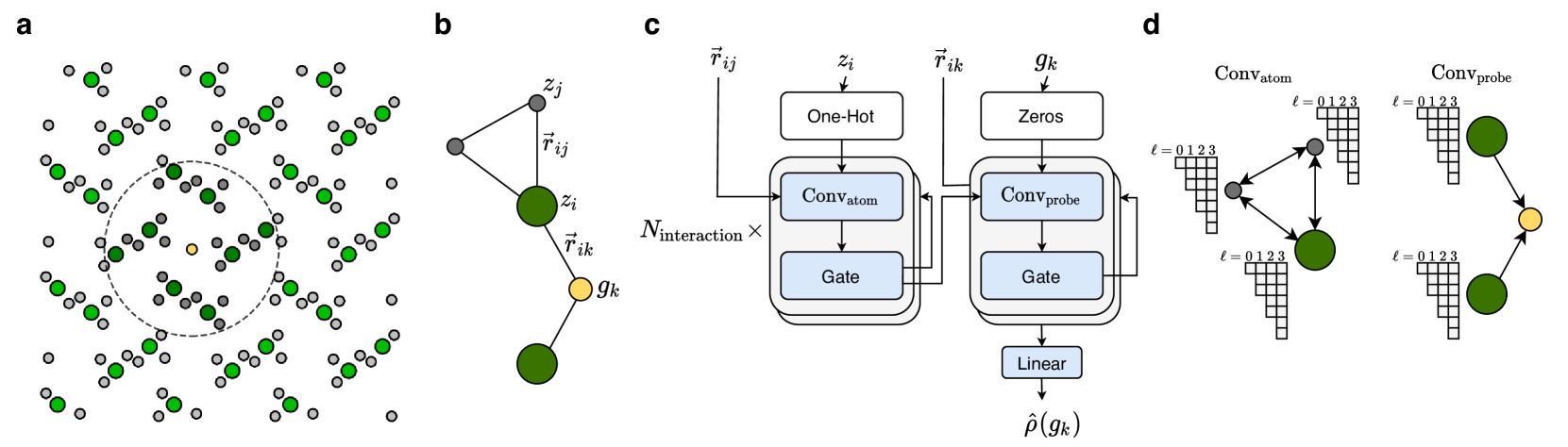
The key innovation of this paper is the development of higher-order equivariant neural networks for predicting charge density in materials. Traditional neural networks for this task often fail to capture the inherent symmetries of atomic structures, leading to suboptimal performance.
To address this, the researchers designed neural network architectures that are equivariant to certain transformations, such as rotations and translations. This means that if the input material is rotated or translated, the neural network's predictions will transform in a corresponding way. The researchers achieved this by introducing higher-order tensor representations that can encode these symmetries.
The performance of the higher-order equivariant neural networks was evaluated on a diverse set of materials, including molecular properties and homogenized metamaterials. The results demonstrate significant improvements in charge density prediction accuracy compared to previous approaches, while also being more computationally efficient.
Critical Analysis
The researchers acknowledge that their approach is limited to materials with well-defined atomic structures, and may not be as effective for more complex or disordered systems. Additionally, the paper does not explore the transferability of the trained models to new materials or the potential for online learning and adaptation.
Further research could investigate ways to extend the equivariant neural network approach to a wider range of materials, including those with defects or amorphous structures. Exploring the integration of SO(3) equivariant representations could also be a promising direction to improve the generalization capabilities of the models.
Conclusion
This paper presents a significant advancement in the field of charge density prediction for materials by introducing higher-order equivariant neural networks. The incorporation of symmetry-aware architectures has led to substantial improvements in accuracy and efficiency, which could have far-reaching implications for materials science and design.
The proposed approach represents an important step towards the development of more accurate and interpretable machine learning models for materials, with potential applications in areas such as catalyst design, molecular property prediction, and metamaterial engineering. As the field of materials informatics continues to evolve, research like this will play a crucial role in unlocking the full potential of computational materials design.
This summary was produced with help from an AI and may contain inaccuracies - check out the links to read the original source documents!
Related Papers
0
Higher-Order Equivariant Neural Networks for Charge Density Prediction in Materials
Teddy Koker, Keegan Quigley, Eric Taw, Kevin Tibbetts, Lin Li
The calculation of electron density distribution using density functional theory (DFT) in materials and molecules is central to the study of their quantum and macro-scale properties, yet accurate and efficient calculation remains a long-standing challenge. We introduce ChargE3Net, an E(3)-equivariant graph neural network for predicting electron density in atomic systems. ChargE3Net enables the learning of higher-order equivariant feature to achieve high predictive accuracy and model expressivity. We show that ChargE3Net exceeds the performance of prior work on diverse sets of molecules and materials. When trained on the massive dataset of over 100K materials in the Materials Project database, our model is able to capture the complexity and variability in the data, leading to a significant 26.7% reduction in self-consistent iterations when used to initialize DFT calculations on unseen materials. Furthermore, we show that non-self-consistent DFT calculations using our predicted charge densities yield near-DFT performance on electronic and thermodynamic property prediction at a fraction of the computational cost. Further analysis attributes the greater predictive accuracy to improved modeling of systems with high angular variations. These results illuminate a pathway towards a machine learning-accelerated ab initio calculations for materials discovery.
Read more5/15/2024

0
A Recipe for Charge Density Prediction
Xiang Fu, Andrew Rosen, Kyle Bystrom, Rui Wang, Albert Musaelian, Boris Kozinsky, Tess Smidt, Tommi Jaakkola
In density functional theory, charge density is the core attribute of atomic systems from which all chemical properties can be derived. Machine learning methods are promising in significantly accelerating charge density prediction, yet existing approaches either lack accuracy or scalability. We propose a recipe that can achieve both. In particular, we identify three key ingredients: (1) representing the charge density with atomic and virtual orbitals (spherical fields centered at atom/virtual coordinates); (2) using expressive and learnable orbital basis sets (basis function for the spherical fields); and (3) using high-capacity equivariant neural network architecture. Our method achieves state-of-the-art accuracy while being more than an order of magnitude faster than existing methods. Furthermore, our method enables flexible efficiency-accuracy trade-offs by adjusting the model/basis sizes.
Read more5/30/2024

0
Highly Accurate Real-space Electron Densities with Neural Networks
Lixue Cheng, P. Bern'at Szab'o, Zeno Schatzle, Derk Kooi, Jonas Kohler, Klaas J. H. Giesbertz, Frank No'e, Jan Hermann, Paola Gori-Giorgi, Adam Foster
Variational ab-initio methods in quantum chemistry stand out among other methods in providing direct access to the wave function. This allows in principle straightforward extraction of any other observable of interest, besides the energy, but in practice this extraction is often technically difficult and computationally impractical. Here, we consider the electron density as a central observable in quantum chemistry and introduce a novel method to obtain accurate densities from real-space many-electron wave functions by representing the density with a neural network that captures known asymptotic properties and is trained from the wave function by score matching and noise-contrastive estimation. We use variational quantum Monte Carlo with deep-learning ansatze (deep QMC) to obtain highly accurate wave functions free of basis set errors, and from them, using our novel method, correspondingly accurate electron densities, which we demonstrate by calculating dipole moments, nuclear forces, contact densities, and other density-based properties.
Read more9/4/2024
🖼️
0
Image Super-resolution Inspired Electron Density Prediction
Chenghan Li, Or Sharir, Shunyue Yuan, Garnet K. Chan
Drawing inspiration from the domain of image super-resolution, we view the electron density as a 3D grayscale image and use a convolutional residual network to transform a crude and trivially generated guess of the molecular density into an accurate ground-state quantum mechanical density. We find that this model outperforms all prior density prediction approaches. Because the input is itself a real-space density, the predictions are equivariant to molecular symmetry transformations even though the model is not constructed to be. Due to its simplicity, the model is directly applicable to unseen molecular conformations and chemical elements. We show that fine-tuning on limited new data provides high accuracy even in challenging cases of exotic elements and charge states. Our work suggests new routes to learning real-space physical quantities drawing from the established ideas of image processing.
Read more8/7/2024