Universal Machine Learning Kohn-Sham Hamiltonian for Materials

0
💬
Sign in to get full access
Overview
- Density Functional Theory (DFT) is a widely used computational approach for electronic structure calculations, but it has limitations in terms of computational demands and scalability.
- Leveraging neural networks to parameterize the Kohn-Sham DFT Hamiltonian has emerged as a promising way to accelerate electronic structure computations.
- However, challenges remain, such as the need for extensive DFT training data and the complexity of establishing accurate machine learning (ML) models for multi-elemental materials.
- This study introduces a universal electronic Hamiltonian model trained on Hamiltonian matrices obtained from first-principles DFT calculations of nearly all crystal structures on the Materials Project.
Plain English Explanation
The paper describes a new approach to modeling the electronic properties of materials using machine learning. Traditionally, Density Functional Theory (DFT) has been used to calculate the electronic structure of materials, but it can be computationally intensive, especially for complex materials.
The researchers in this study have developed a "universal electronic Hamiltonian model" that can predict the electronic properties of a wide range of materials, including complex multi-element systems, solid-state electrolytes, and metal-organic frameworks (MOFs). This model is trained on a large dataset of Hamiltonian matrices, which are mathematical representations of the electronic structure, obtained from DFT calculations.
By using this universal model, the researchers were able to quickly calculate the electronic properties of thousands of materials, identifying those with useful properties like direct band gaps and flat bands. This could accelerate the discovery and design of new materials for applications like energy storage, electronics, and catalysis.
Technical Explanation
The researchers developed a universal electronic Hamiltonian model trained on Hamiltonian matrices obtained from first-principles DFT calculations of nearly all crystal structures in the Materials Project database. This allowed them to create a model capable of accurately predicting electronic structures across the entire periodic table, including complex multi-elemental systems.
The model was tested on a variety of material systems, including solid-state electrolytes, Moiré twisted bilayer heterostructures, and metal-organic frameworks (MOFs). The researchers were able to use the universal model to conduct high-throughput calculations of electronic structures for over 100,000 crystals in the GeNOME dataset, identifying thousands with potentially useful properties like direct band gaps and flat bands.
This approach addresses some of the key challenges in applying machine learning to electronic structure calculations, such as the need for extensive DFT training data and the difficulty of building accurate models for multi-elemental materials. By leveraging a large, diverse dataset and a universal Hamiltonian model, the researchers have created a reliable, efficient framework for computing electronic properties that could enable advancements in fields like materials design and discovery.
Critical Analysis
The researchers have made significant progress in addressing the limitations of traditional DFT approaches by developing a universal electronic Hamiltonian model. This model's ability to accurately predict electronic structures across a wide range of materials, including complex multi-elemental systems, is a notable achievement.
However, the paper does not extensively discuss the model's limitations or potential areas for further improvement. For example, it would be valuable to understand the model's performance on materials with exotic or unusual electronic properties, or to explore ways to further optimize its computational efficiency.
Additionally, while the researchers demonstrate the model's ability to identify materials with desirable electronic properties, such as direct band gaps and flat bands, they do not provide a detailed analysis of the practical implications or potential applications of these findings. It would be helpful to see a more in-depth discussion of how these materials could be used in real-world technologies and the potential impact on fields like energy storage, electronics, and catalysis.
Overall, this research represents an important step forward in the development of data-driven approaches for electronic structure calculations. By creating a universal, efficient model, the researchers have laid the groundwork for accelerating materials discovery and design across a wide range of applications.
Conclusion
This study introduces a novel universal electronic Hamiltonian model that can accurately predict the electronic properties of a diverse range of materials, including complex multi-elemental systems. By leveraging a large dataset of Hamiltonian matrices from first-principles DFT calculations, the researchers have created a reliable and efficient framework for computing electronic structures.
The ability to quickly identify materials with desirable properties, such as direct band gaps and flat bands, could have significant implications for fields like energy storage, electronics, and catalysis. This research represents an important step forward in the development of data-driven approaches to materials discovery and design, laying the groundwork for accelerating the development of new technologies.
This summary was produced with help from an AI and may contain inaccuracies - check out the links to read the original source documents!
Related Papers
💬
0
Universal Machine Learning Kohn-Sham Hamiltonian for Materials
Yang Zhong, Hongyu Yu, Jihui Yang, Xingyu Guo, Hongjun Xiang, Xingao Gong
While density functional theory (DFT) serves as a prevalent computational approach in electronic structure calculations, its computational demands and scalability limitations persist. Recently, leveraging neural networks to parameterize the Kohn-Sham DFT Hamiltonian has emerged as a promising avenue for accelerating electronic structure computations. Despite advancements, challenges such as the necessity for computing extensive DFT training data to explore each new system and the complexity of establishing accurate ML models for multi-elemental materials still exist. Addressing these hurdles, this study introduces a universal electronic Hamiltonian model trained on Hamiltonian matrices obtained from first-principles DFT calculations of nearly all crystal structures on the Materials Project. We demonstrate its generality in predicting electronic structures across the whole periodic table, including complex multi-elemental systems, solid-state electrolytes, Moir'e twisted bilayer heterostructure, and metal-organic frameworks (MOFs). Moreover, we utilize the universal model to conduct high-throughput calculations of electronic structures for crystals in GeNOME datasets, identifying 3,940 crystals with direct band gaps and 5,109 crystals with flat bands. By offering a reliable efficient framework for computing electronic properties, this universal Hamiltonian model lays the groundwork for advancements in diverse fields, such as easily providing a huge data set of electronic structures and also making the materials design across the whole periodic table possible.
Read more4/16/2024
0
Machine learning Hubbard parameters with equivariant neural networks
Martin Uhrin, Austin Zadoks, Luca Binci, Nicola Marzari, Iurii Timrov
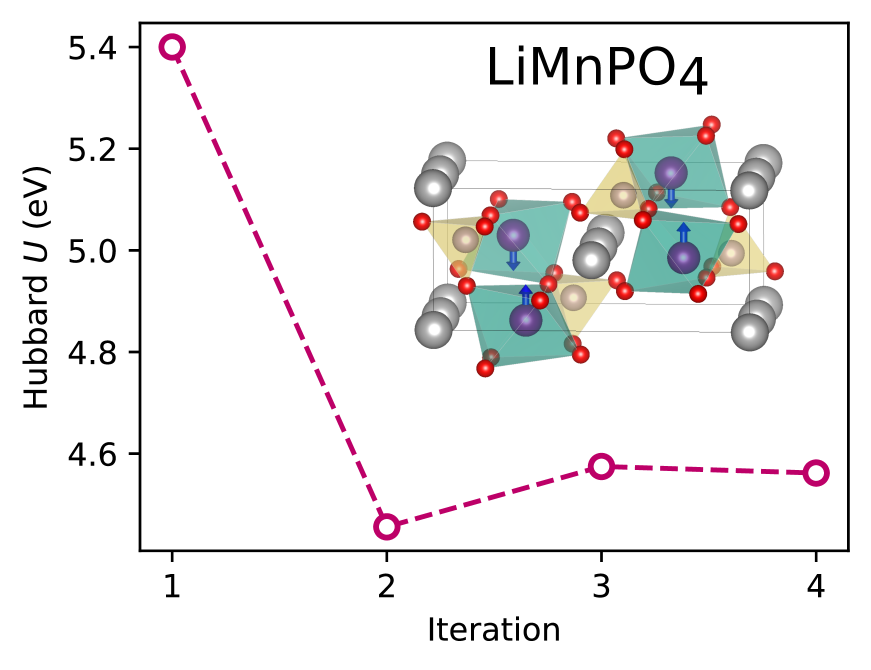
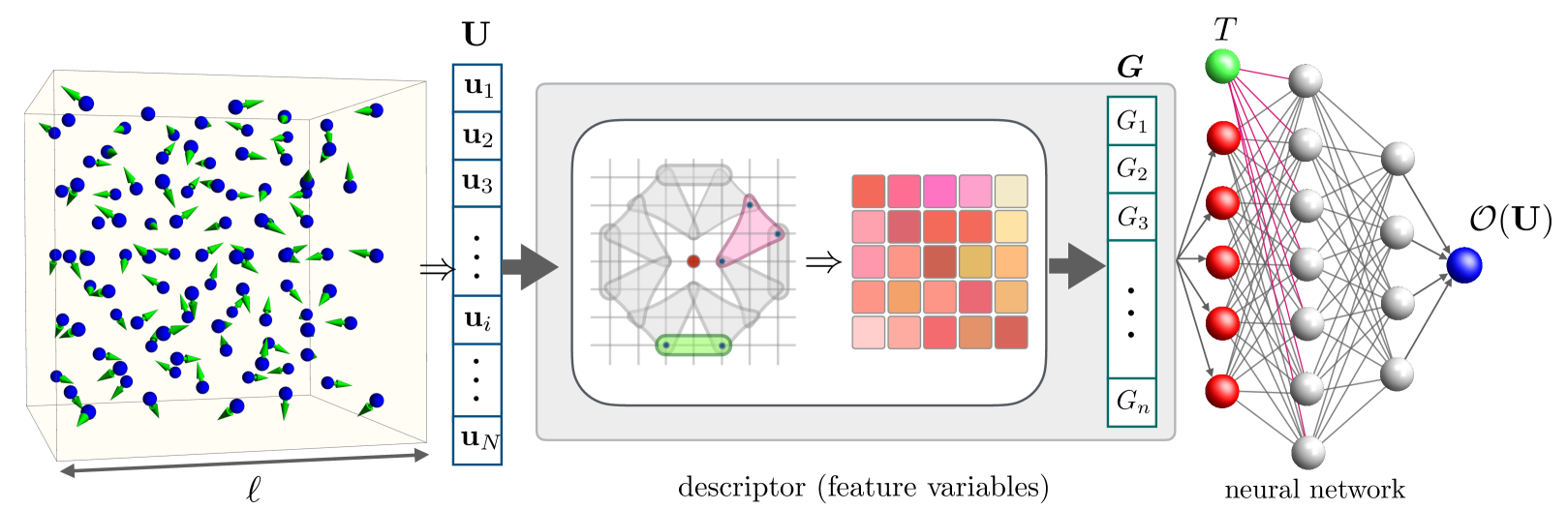
Density-functional theory with extended Hubbard functionals (DFT+$U$+$V$) provides a robust framework to accurately describe complex materials containing transition-metal or rare-earth elements. It does so by mitigating self-interaction errors inherent to semi-local functionals which are particularly pronounced in systems with partially-filled $d$ and $f$ electronic states. However, achieving accuracy in this approach hinges upon the accurate determination of the on-site $U$ and inter-site $V$ Hubbard parameters. In practice, these are obtained either by semi-empirical tuning, requiring prior knowledge, or, more correctly, by using predictive but expensive first-principles calculations. Here, we present a machine learning model based on equivariant neural networks which uses atomic occupation matrices as descriptors, directly capturing the electronic structure, local chemical environment, and oxidation states of the system at hand. We target here the prediction of Hubbard parameters computed self-consistently with iterative linear-response calculations, as implemented in density-functional perturbation theory (DFPT), and structural relaxations. Remarkably, when trained on data from 11 materials spanning various crystal structures and compositions, our model achieves mean absolute relative errors of 3% and 5% for Hubbard $U$ and $V$ parameters, respectively. By circumventing computationally expensive DFT or DFPT self-consistent protocols, our model significantly expedites the prediction of Hubbard parameters with negligible computational overhead, while approaching the accuracy of DFPT. Moreover, owing to its robust transferability, the model facilitates accelerated materials discovery and design via high-throughput calculations, with relevance for various technological applications.
Read more6/5/2024
🎯
0
Multi-task learning for molecular electronic structure approaching coupled-cluster accuracy
Hao Tang, Brian Xiao, Wenhao He, Pero Subasic, Avetik R. Harutyunyan, Yao Wang, Fang Liu, Haowei Xu, Ju Li
Machine learning (ML) plays an important role in quantum chemistry, providing fast-to-evaluate predictive models for various properties of molecules. However, as most existing ML models for molecular electronic properties use density function theory (DFT) databases as the ground truth in training, their prediction accuracy cannot go beyond the DFT. In this work, we developed a unified ML method for electronic structures of organic molecules using the gold-standard CCSD(T) calculations as training data. Tested on hydrocarbon molecules, our model outperforms the DFT with the widely-used B3LYP functional in both computation costs and prediction accuracy of various quantum chemical properties. We apply the model to aromatic compounds and semiconducting polymers on both ground state and excited state properties, demonstrating its accuracy and generalization capability to complex systems that are hard to calculate using CCSD(T)-level methods.
Read more5/22/2024
0
Machine learning approach for vibronically renormalized electronic band structures
Niraj Aryal, Sheng Zhang, Weiguo Yin, Gia-Wei Chern
We present a machine learning (ML) method for efficient computation of vibrational thermal expectation values of physical properties from first principles. Our approach is based on the non-perturbative frozen phonon formulation in which stochastic Monte Carlo algorithm is employed to sample configurations of nuclei in a supercell at finite temperatures based on a first-principles phonon model. A deep-learning neural network is trained to accurately predict physical properties associated with sampled phonon configurations, thus bypassing the time-consuming {em ab initio} calculations. To incorporate the point-group symmetry of the electronic system into the ML model, group-theoretical methods are used to develop a symmetry-invariant descriptor for phonon configurations in the supercell. We apply our ML approach to compute the temperature dependent electronic energy gap of silicon based on density functional theory (DFT). We show that, with less than a hundred DFT calculations for training the neural network model, an order of magnitude larger number of sampling can be achieved for the computation of the vibrational thermal expectation values. Our work highlights the promising potential of ML techniques for finite temperature first-principles electronic structure methods.
Read more9/4/2024