A Hitchhiker's Guide to Deep Chemical Language Processing for Bioactivity Prediction

0

Sign in to get full access
Overview
- This paper explores the use of deep learning language models for predicting the bioactivity of chemical compounds.
- The authors develop a framework called Deep Chemical Language Processing (DCLP) that leverages transformer-based models to encode chemical structures and predict their biological activities.
- The DCLP approach is shown to outperform traditional cheminformatics methods on several benchmark datasets for drug discovery and development.
Plain English Explanation
The paper describes a new way to analyze and understand the properties of chemical compounds using deep learning language models. These models are inspired by the way humans process and understand natural language, but instead of working with words, they operate on the building blocks of chemical structures.
The key idea is to represent the chemical structure of a compound as a "sentence" made up of the various atoms and bonds that make up the molecule. Just as language models can learn patterns and meanings from large text datasets, the DCLP framework can learn the underlying relationships between chemical structure and biological activity by training on large datasets of known compounds and their effects.
This allows the model to make predictions about the potential bioactivity of new, untested compounds - which is crucial for accelerating drug discovery and development. The authors demonstrate that their DCLP approach outperforms traditional cheminformatics methods, which rely more on manual feature engineering and rule-based systems.
By treating chemicals like a language that can be learned and understood by deep learning models, the researchers have opened up new possibilities for harnessing the power of artificial intelligence to navigate the vast chemical space and uncover promising drug candidates more efficiently.
Technical Explanation
The paper introduces a framework called Deep Chemical Language Processing (DCLP) that adapts transformer-based language models to the task of predicting the bioactivity of chemical compounds. The authors represent the molecular structure of a compound as a "SMILES string" - a textual encoding of the atoms and bonds - and feed this into a transformer model.
The transformer model learns to extract relevant features and patterns from the chemical "sentences," allowing it to predict the biological activity of the compound (e.g., whether it is likely to be a useful drug). The DCLP framework is evaluated on several standard benchmarks for drug discovery, including tasks like toxicity prediction, protein-ligand binding affinity estimation, and activity classification.
The results show that the DCLP approach outperforms traditional cheminformatics methods that rely on manual feature engineering and rule-based systems. The authors attribute this to the ability of the transformer model to automatically learn complex representations of chemical structure and discover meaningful patterns from the data.
Additionally, the paper explores the interpretability of the DCLP model, demonstrating how the attention mechanism can be used to identify the most important substructures contributing to the predicted bioactivity. This provides valuable insights that can guide further experimental work and hypothesis generation.
Critical Analysis
The paper presents a compelling approach to leveraging deep learning for chemical bioactivity prediction, and the results clearly demonstrate the potential of this technique. However, it's important to consider some of the potential limitations and areas for further research:
-
The DCLP framework relies on the availability of large, high-quality datasets of chemical structures and their associated bioactivities. In practice, such comprehensive data may not always be readily available, particularly for more specialized or niche applications.
-
While the attention mechanism provides some interpretability, the inner workings of the transformer model can still be opaque. Further research is needed to improve the transparency and explainability of these models, which is crucial for building trust and acceptance in the drug discovery community.
-
The paper focuses on predicting bioactivity based on chemical structure alone, but in reality, the biological activity of a compound is influenced by a complex interplay of factors, including pharmacokinetics, toxicology, and target interactions. Integrating these additional layers of information could further improve the predictive power of the DCLP approach.
-
As with any machine learning model, the DCLP framework is susceptible to bias and may struggle to generalize well to completely novel chemical space. Continued efforts to robustly evaluate and validate these models are necessary to ensure their reliability and applicability.
Conclusion
The "Hitchhiker's Guide to Deep Chemical Language Processing for Bioactivity Prediction" presents a novel and promising approach to leveraging deep learning for accelerating drug discovery. By treating chemical compounds as a "language" that can be learned and understood by transformer-based models, the DCLP framework demonstrates significant improvements over traditional cheminformatics methods.
The ability to accurately predict the bioactivity of chemical compounds based on their structural features alone has the potential to dramatically streamline the drug discovery process, helping researchers identify promising drug candidates more efficiently and ultimately improve patient outcomes. As the field of AI-powered drug discovery continues to evolve, the insights and techniques presented in this paper are likely to play an increasingly important role.
This summary was produced with help from an AI and may contain inaccuracies - check out the links to read the original source documents!
Related Papers
0
A Hitchhiker's Guide to Deep Chemical Language Processing for Bioactivity Prediction
R{i}za Ozc{c}elik, Francesca Grisoni
Deep learning has significantly accelerated drug discovery, with 'chemical language' processing (CLP) emerging as a prominent approach. CLP learns from molecular string representations (e.g., Simplified Molecular Input Line Entry Systems [SMILES] and Self-Referencing Embedded Strings [SELFIES]) with methods akin to natural language processing. Despite their growing importance, training predictive CLP models is far from trivial, as it involves many 'bells and whistles'. Here, we analyze the key elements of CLP training, to provide guidelines for newcomers and experts alike. Our study spans three neural network architectures, two string representations, three embedding strategies, across ten bioactivity datasets, for both classification and regression purposes. This 'hitchhiker's guide' not only underscores the importance of certain methodological choices, but it also equips researchers with practical recommendations on ideal choices, e.g., in terms of neural network architectures, molecular representations, and hyperparameter optimization.
Read more7/18/2024
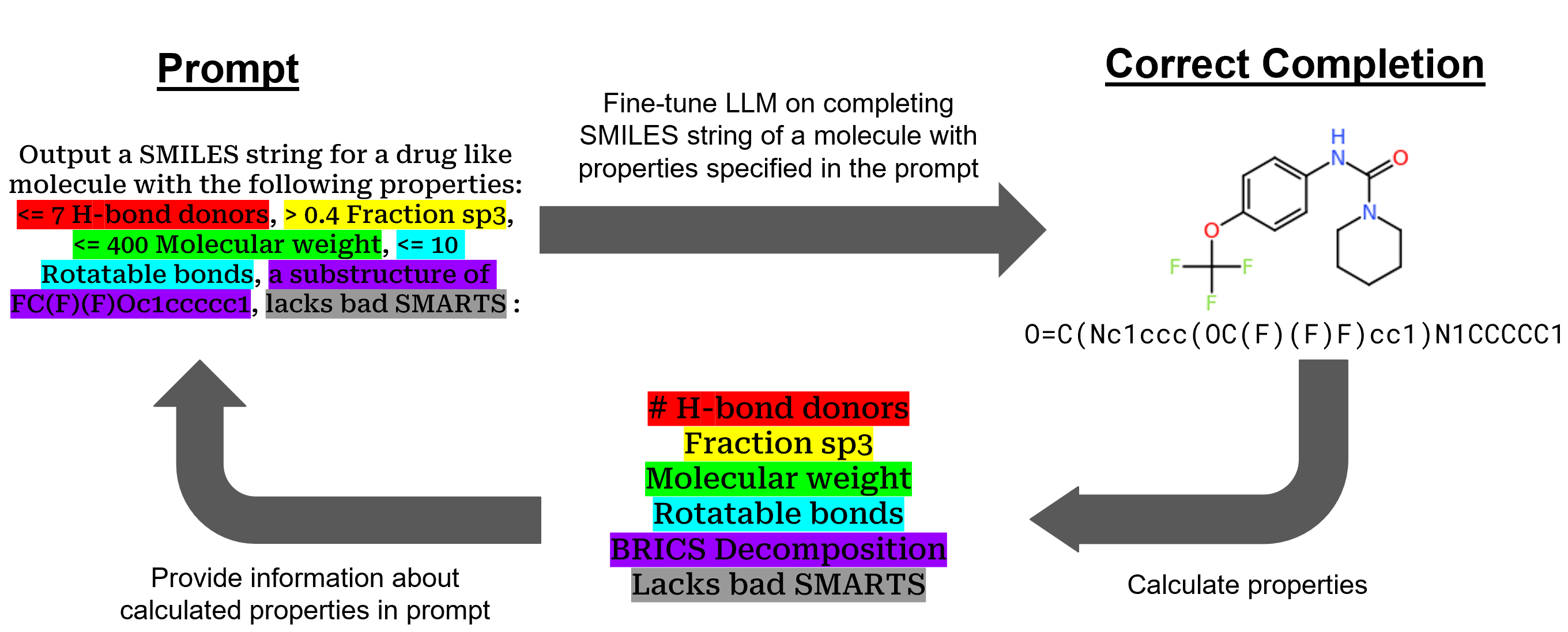
0
SmileyLlama: Modifying Large Language Models for Directed Chemical Space Exploration
Joseph M. Cavanagh, Kunyang Sun, Andrew Gritsevskiy, Dorian Bagni, Thomas D. Bannister, Teresa Head-Gordon
Here we show that a Large Language Model (LLM) can serve as a foundation model for a Chemical Language Model (CLM) which performs at or above the level of CLMs trained solely on chemical SMILES string data. Using supervised fine-tuning (SFT) and direct preference optimization (DPO) on the open-source Llama LLM, we demonstrate that we can train an LLM to respond to prompts such as generating molecules with properties of interest to drug development. This overall framework allows an LLM to not just be a chatbot client for chemistry and materials tasks, but can be adapted to speak more directly as a CLM which can generate molecules with user-specified properties.
Read more9/12/2024

0
Active Causal Learning for Decoding Chemical Complexities with Targeted Interventions
Zachary R. Fox, Ayana Ghosh
Predicting and enhancing inherent properties based on molecular structures is paramount to design tasks in medicine, materials science, and environmental management. Most of the current machine learning and deep learning approaches have become standard for predictions, but they face challenges when applied across different datasets due to reliance on correlations between molecular representation and target properties. These approaches typically depend on large datasets to capture the diversity within the chemical space, facilitating a more accurate approximation, interpolation, or extrapolation of the chemical behavior of molecules. In our research, we introduce an active learning approach that discerns underlying cause-effect relationships through strategic sampling with the use of a graph loss function. This method identifies the smallest subset of the dataset capable of encoding the most information representative of a much larger chemical space. The identified causal relations are then leveraged to conduct systematic interventions, optimizing the design task within a chemical space that the models have not encountered previously. While our implementation focused on the QM9 quantum-chemical dataset for a specific design task-finding molecules with a large dipole moment-our active causal learning approach, driven by intelligent sampling and interventions, holds potential for broader applications in molecular, materials design and discovery.
Read more4/8/2024

0
MolTailor: Tailoring Chemical Molecular Representation to Specific Tasks via Text Prompts
Haoqiang Guo, Sendong Zhao, Haochun Wang, Yanrui Du, Bing Qin
Deep learning is now widely used in drug discovery, providing significant acceleration and cost reduction. As the most fundamental building block, molecular representation is essential for predicting molecular properties to enable various downstream applications. Most existing methods attempt to incorporate more information to learn better representations. However, not all features are equally important for a specific task. Ignoring this would potentially compromise the training efficiency and predictive accuracy. To address this issue, we propose a novel approach, which treats language models as an agent and molecular pretraining models as a knowledge base. The agent accentuates task-relevant features in the molecular representation by understanding the natural language description of the task, just as a tailor customizes clothes for clients. Thus, we call this approach MolTailor. Evaluations demonstrate MolTailor's superior performance over baselines, validating the efficacy of enhancing relevance for molecular representation learning. This illustrates the potential of language model guided optimization to better exploit and unleash the capabilities of existing powerful molecular representation methods. Our code is available at https://github.com/SCIR-HI/MolTailor.
Read more4/22/2024