Kinematics Modeling of Peroxy Free Radicals: A Deep Reinforcement Learning Approach

0
Sign in to get full access
Overview
- This paper presents a deep reinforcement learning approach for modeling the kinematics of peroxy free radicals, which are important intermediates in atmospheric chemistry and combustion processes.
- The researchers developed a neural network-based model that can accurately predict the behavior of these free radicals without relying on complex physical simulations.
- The model was trained on a large dataset of quantum chemical calculations and was able to outperform traditional regression methods in terms of accuracy and computational efficiency.
Plain English Explanation
Free radicals are unstable molecules with unpaired electrons, which makes them highly reactive. Peroxy free radicals are a specific type of free radical that play a crucial role in atmospheric chemistry and combustion reactions. Understanding the behavior of these radicals is important for accurately modeling and predicting complex chemical processes.
In this paper, the researchers used a deep learning approach to model the kinematics, or the motion and behavior, of peroxy free radicals. Instead of relying on complex physical simulations, they trained a neural network model on a large dataset of quantum chemical calculations. This allowed the model to learn the underlying patterns and relationships in the data, enabling it to accurately predict the behavior of the free radicals without the need for computationally intensive simulations.
The key advantage of this approach is that it is much more efficient than traditional methods, while still maintaining a high level of accuracy. This could have important implications for fields like atmospheric chemistry and combustion research, where the ability to quickly and accurately model complex chemical processes is crucial.
Technical Explanation
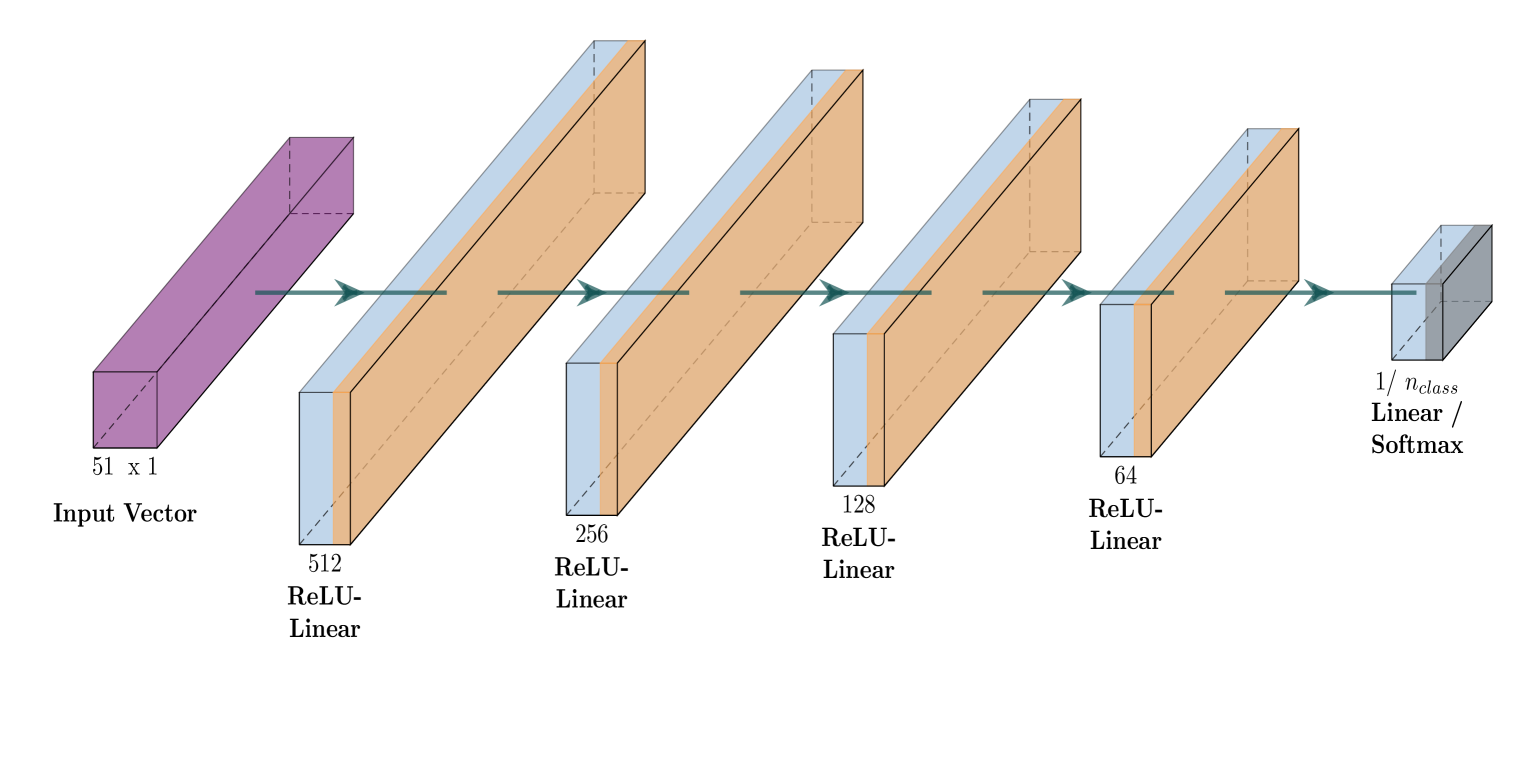
The researchers employed a deep reinforcement learning approach to develop a neural network model for predicting the kinematics of peroxy free radicals. They first generated a large dataset of quantum chemical calculations, which provided the ground truth information about the behavior of these free radicals.
The neural network model was then trained on this dataset using a reinforcement learning algorithm. The model was designed to take in the chemical structure and environmental conditions of the free radical as input, and output predictions about its kinematic behavior, such as its rotational and translational motion.
The researchers compared the performance of their deep learning model to traditional regression-based methods, and found that their approach outperformed the traditional methods in terms of both accuracy and computational efficiency. This was due to the model's ability to capture the complex nonlinear relationships in the data, which are difficult for traditional methods to model effectively.
Critical Analysis
The researchers acknowledge several limitations of their work, including the fact that the dataset used for training the model was limited to a specific set of conditions and chemical environments. This means that the model's performance may not generalize well to more complex or diverse scenarios.
Additionally, the paper does not provide a detailed analysis of the model's interpretability or the extent to which the learned representations align with our understanding of the underlying physical processes. This is an important consideration, as the ability to understand and explain the model's predictions can be crucial for building trust and confidence in the technology.
Despite these limitations, the researchers have demonstrated the potential of deep reinforcement learning for modeling the kinematics of peroxy free radicals. Their work highlights the value of leveraging large datasets and powerful machine learning algorithms to tackle complex problems in chemistry and physics that have traditionally been the domain of complex physical simulations.
Conclusion
This paper presents a novel deep reinforcement learning approach for modeling the kinematics of peroxy free radicals, which are important intermediates in atmospheric chemistry and combustion processes. The researchers developed a neural network-based model that can accurately predict the behavior of these free radicals without relying on computationally intensive physical simulations.
The key advantage of this approach is its efficiency and scalability, which could have important implications for fields like atmospheric chemistry and combustion research. While the model has some limitations, the researchers have demonstrated the potential of deep learning for tackling complex problems in chemistry and physics, and their work paves the way for further advancements in this area.
This summary was produced with help from an AI and may contain inaccuracies - check out the links to read the original source documents!
Related Papers
0
Kinematics Modeling of Peroxy Free Radicals: A Deep Reinforcement Learning Approach
Subhadarsi Nayak, Hrithwik Shalu, Joseph Stember
Tropospheric ozone, known as a concerning air pollutant, has been associated with health issues including asthma, bronchitis, and impaired lung function. The rates at which peroxy radicals react with NO play a critical role in the overall formation and depletion of tropospheric ozone. However, obtaining comprehensive kinetic data for these reactions remains challenging. Traditional approaches to determine rate constants are costly and technically intricate. Fortunately, the emergence of machine learning-based models offers a less resource and time-intensive alternative for acquiring kinetics information. In this study, we leveraged deep reinforcement learning to predict ranges of rate constants (textit{k}) with exceptional accuracy, achieving a testing set accuracy of 100%. To analyze reactivity trends based on the molecular structure of peroxy radicals, we employed 51 global descriptors as input parameters. These descriptors were derived from optimized minimum energy geometries of peroxy radicals using the quantum composite G3B3 method. Through the application of Integrated Gradients (IGs), we gained valuable insights into the significance of the various descriptors in relation to reaction rates. We successfully validated and contextualized our findings by conducting cross-comparisons with established trends in the existing literature. These results establish a solid foundation for pioneering advancements in chemistry, where computer analysis serves as an inspirational source driving innovation.
Read more4/17/2024

0
3DReact: Geometric deep learning for chemical reactions
Puck van Gerwen, Ksenia R. Briling, Charlotte Bunne, Vignesh Ram Somnath, Ruben Laplaza, Andreas Krause, Clemence Corminboeuf
Geometric deep learning models, which incorporate the relevant molecular symmetries within the neural network architecture, have considerably improved the accuracy and data efficiency of predictions of molecular properties. Building on this success, we introduce 3DReact, a geometric deep learning model to predict reaction properties from three-dimensional structures of reactants and products. We demonstrate that the invariant version of the model is sufficient for existing reaction datasets. We illustrate its competitive performance on the prediction of activation barriers on the GDB7-22-TS, Cyclo-23-TS and Proparg-21-TS datasets in different atom-mapping regimes. We show that, compared to existing models for reaction property prediction, 3DReact offers a flexible framework that exploits atom-mapping information, if available, as well as geometries of reactants and products (in an invariant or equivariant fashion). Accordingly, it performs systematically well across different datasets, atom-mapping regimes, as well as both interpolation and extrapolation tasks.
Read more7/15/2024

0
Towards Foundation Models for the Industrial Forecasting of Chemical Kinetics
Imran Nasim, Joa~o Lucas de Sousa Almeida
Scientific Machine Learning is transforming traditional engineering industries by enhancing the efficiency of existing technologies and accelerating innovation, particularly in modeling chemical reactions. Despite recent advancements, the issue of solving stiff chemically reacting problems within computational fluid dynamics remains a significant issue. In this study we propose a novel approach utilizing a multi-layer-perceptron mixer architecture (MLP-Mixer) to model the time-series of stiff chemical kinetics. We evaluate this method using the ROBER system, a benchmark model in chemical kinetics, to compare its performance with traditional numerical techniques. This study provides insight into the industrial utility of the recently developed MLP-Mixer architecture to model chemical kinetics and provides motivation for such neural architecture to be used as a base for time-series foundation models.
Read more8/21/2024

0
ReactAIvate: A Deep Learning Approach to Predicting Reaction Mechanisms and Unmasking Reactivity Hotspots
Ajnabiul Hoque, Manajit Das, Mayank Baranwal, Raghavan B. Sunoj
A chemical reaction mechanism (CRM) is a sequence of molecular-level events involving bond-breaking/forming processes, generating transient intermediates along the reaction pathway as reactants transform into products. Understanding such mechanisms is crucial for designing and discovering new reactions. One of the currently available methods to probe CRMs is quantum mechanical (QM) computations. The resource-intensive nature of QM methods and the scarcity of mechanism-based datasets motivated us to develop reliable ML models for predicting mechanisms. In this study, we created a comprehensive dataset with seven distinct classes, each representing uniquely characterized elementary steps. Subsequently, we developed an interpretable attention-based GNN that achieved near-unity and 96% accuracy, respectively for reaction step classification and the prediction of reactive atoms in each such step, capturing interactions between the broader reaction context and local active regions. The near-perfect classification enables accurate prediction of both individual events and the entire CRM, mitigating potential drawbacks of Seq2Seq approaches, where a wrongly predicted character leads to incoherent CRM identification. In addition to interpretability, our model adeptly identifies key atom(s) even from out-of-distribution classes. This generalizabilty allows for the inclusion of new reaction types in a modular fashion, thus will be of value to experts for understanding the reactivity of new molecules.
Read more7/16/2024