ReactAIvate: A Deep Learning Approach to Predicting Reaction Mechanisms and Unmasking Reactivity Hotspots

0

Sign in to get full access
Overview
- This paper presents a deep learning approach called ReactAIvate for predicting reaction mechanisms and identifying reactivity hotspots.
- The method uses advanced neural network architectures to analyze chemical structures and reaction conditions to forecast the likely reaction pathways and key reactive sites.
- The research aims to provide a powerful tool for accelerating the discovery and optimization of new chemical reactions, with potential applications in areas like drug development, materials science, and sustainable chemistry.
Plain English Explanation
The paper describes a new artificial intelligence (AI) system called ReactAIvate that can help chemists understand and predict how chemical reactions work. When chemists are trying to develop a new chemical process, a key challenge is figuring out the exact sequence of steps, or "mechanism," that the reaction follows. ReactAIvate uses deep learning, a type of AI that can analyze complex patterns in data, to analyze the structure of chemical reactants and the conditions of the reaction. Based on this analysis, the system can forecast the most likely reaction pathway and identify the specific atoms or regions that are the most "reactive" - meaning they are the key sites where the critical chemical transformations occur.
By providing these insights, ReactAIvate aims to accelerate the discovery and optimization of new chemical reactions, which has important applications in fields like drug development, creating new materials, and designing greener, more sustainable chemical processes. The researchers demonstrate how ReactAIvate can outperform previous computational approaches for predicting reaction mechanisms and pinpointing reactivity hotspots. Overall, this AI system represents an exciting advance that could significantly enhance chemists' ability to innovate and solve complex challenges.
Technical Explanation
The core of the ReactAIvate approach is a deep neural network architecture that takes as input the molecular structures of the reactants, the reaction conditions (e.g., temperature, solvent), and other contextual information. The network is trained on a large dataset of known chemical reactions to learn the underlying patterns that govern reaction mechanisms and reactivity.
The key innovations in the neural network design include:
- A specialized graph neural network that can effectively encode the 3D structure and atomic connectivity of the molecules.
- Attention mechanisms that allow the model to focus on the most relevant molecular features when making predictions.
- Causal reasoning modules that can infer the likely causal relationships between reaction conditions, structural changes, and product formation.
The researchers evaluate ReactAIvate on a range of benchmark datasets for reaction mechanism prediction and reactivity hotspot identification. The results demonstrate significant performance gains over previous state-of-the-art methods, such as those based on rule-based expert systems or less sophisticated machine learning approaches.
Critical Analysis
The authors acknowledge several limitations of the current ReactAIvate system. For example, the model is trained on a finite dataset of reactions, so its performance may be constrained by the coverage and quality of the training data. There are also challenges in accurately modeling the complex quantum mechanical and kinetic effects that govern chemical reactivity.
Additionally, while ReactAIvate can identify key reactive sites, the system does not provide a complete mechanistic explanation for why certain atoms or regions are more reactive. Further research would be needed to fully unpack the causal relationships and physical principles underlying the model's predictions.
Despite these caveats, the ReactAIvate approach represents an important step forward in applying deep learning to problems in synthetic chemistry. By continuing to advance these types of AI-powered tools, researchers may be able to unlock new pathways for molecular design and discovery that were previously inaccessible. Critical assessment and further validation of the system's capabilities will be important as the technology matures.
Conclusion
The ReactAIvate deep learning framework offers a promising new approach for predicting reaction mechanisms and identifying reactivity hotspots in chemical systems. By leveraging advanced neural network architectures, the system can analyze complex structural and contextual information to generate insights that could accelerate the development of new chemical processes and products.
While there are still some limitations to the current implementation, the demonstrated performance gains over prior methods suggest that AI-powered tools like ReactAIvate have immense potential to transform how chemists approach problems. As the field of computational reaction modeling continues to evolve, these types of intelligent systems may become indispensable assistants in the quest to unlock the mysteries of chemical reactivity and harness them for the benefit of society.
This summary was produced with help from an AI and may contain inaccuracies - check out the links to read the original source documents!
Related Papers
0
ReactAIvate: A Deep Learning Approach to Predicting Reaction Mechanisms and Unmasking Reactivity Hotspots
Ajnabiul Hoque, Manajit Das, Mayank Baranwal, Raghavan B. Sunoj
A chemical reaction mechanism (CRM) is a sequence of molecular-level events involving bond-breaking/forming processes, generating transient intermediates along the reaction pathway as reactants transform into products. Understanding such mechanisms is crucial for designing and discovering new reactions. One of the currently available methods to probe CRMs is quantum mechanical (QM) computations. The resource-intensive nature of QM methods and the scarcity of mechanism-based datasets motivated us to develop reliable ML models for predicting mechanisms. In this study, we created a comprehensive dataset with seven distinct classes, each representing uniquely characterized elementary steps. Subsequently, we developed an interpretable attention-based GNN that achieved near-unity and 96% accuracy, respectively for reaction step classification and the prediction of reactive atoms in each such step, capturing interactions between the broader reaction context and local active regions. The near-perfect classification enables accurate prediction of both individual events and the entire CRM, mitigating potential drawbacks of Seq2Seq approaches, where a wrongly predicted character leads to incoherent CRM identification. In addition to interpretability, our model adeptly identifies key atom(s) even from out-of-distribution classes. This generalizabilty allows for the inclusion of new reaction types in a modular fashion, thus will be of value to experts for understanding the reactivity of new molecules.
Read more7/16/2024
0
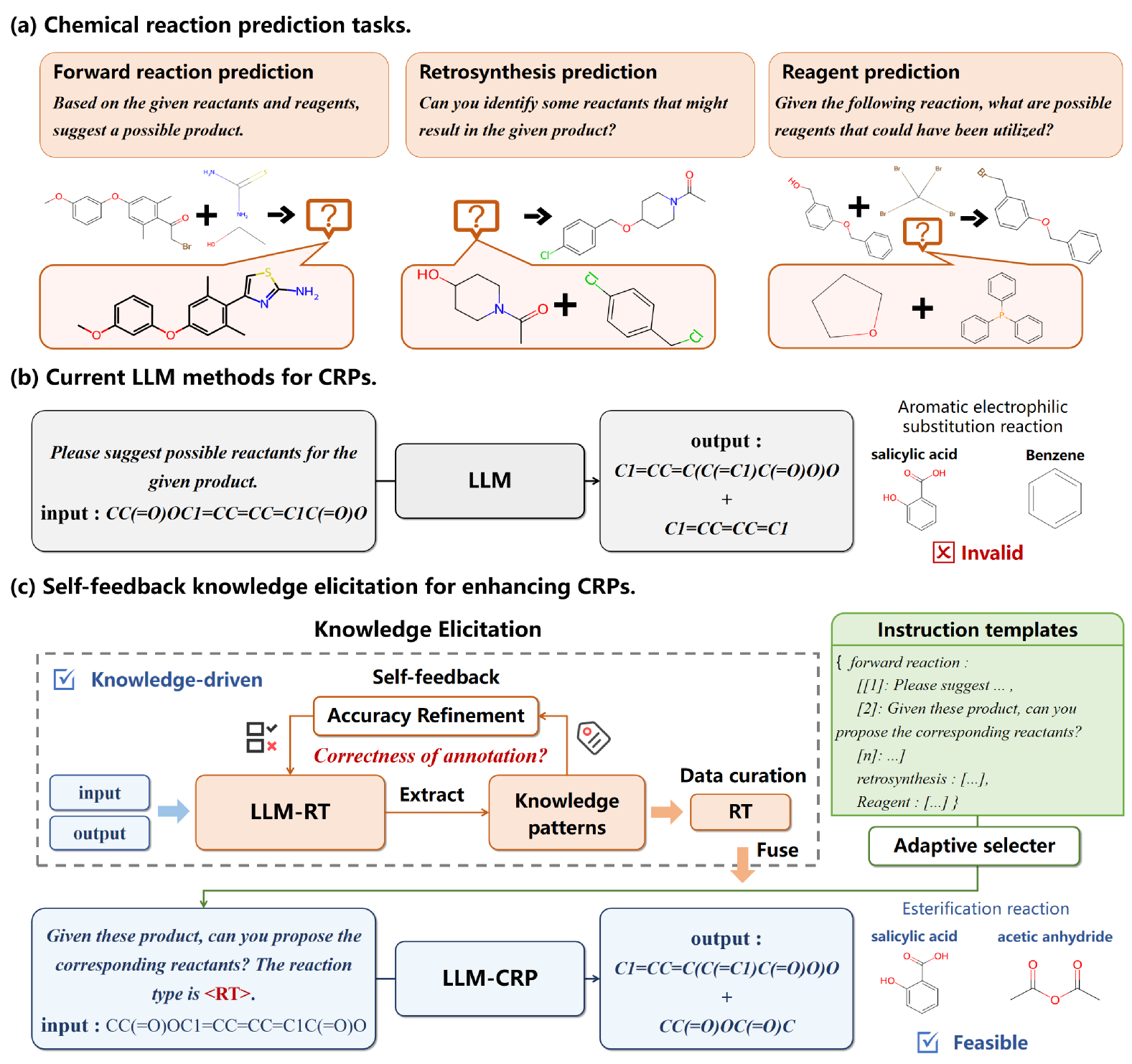
A Self-feedback Knowledge Elicitation Approach for Chemical Reaction Predictions
Pengfei Liu, Jun Tao, Zhixiang Ren
The task of chemical reaction predictions (CRPs) plays a pivotal role in advancing drug discovery and material science. However, its effectiveness is constrained by the vast and uncertain chemical reaction space and challenges in capturing reaction selectivity, particularly due to existing methods' limitations in exploiting the data's inherent knowledge. To address these challenges, we introduce a data-curated self-feedback knowledge elicitation approach. This method starts from iterative optimization of molecular representations and facilitates the extraction of knowledge on chemical reaction types (RTs). Then, we employ adaptive prompt learning to infuse the prior knowledge into the large language model (LLM). As a result, we achieve significant enhancements: a 14.2% increase in retrosynthesis prediction accuracy, a 74.2% rise in reagent prediction accuracy, and an expansion in the model's capability for handling multi-task chemical reactions. This research offers a novel paradigm for knowledge elicitation in scientific research and showcases the untapped potential of LLMs in CRPs.
Read more4/16/2024

0
3DReact: Geometric deep learning for chemical reactions
Puck van Gerwen, Ksenia R. Briling, Charlotte Bunne, Vignesh Ram Somnath, Ruben Laplaza, Andreas Krause, Clemence Corminboeuf
Geometric deep learning models, which incorporate the relevant molecular symmetries within the neural network architecture, have considerably improved the accuracy and data efficiency of predictions of molecular properties. Building on this success, we introduce 3DReact, a geometric deep learning model to predict reaction properties from three-dimensional structures of reactants and products. We demonstrate that the invariant version of the model is sufficient for existing reaction datasets. We illustrate its competitive performance on the prediction of activation barriers on the GDB7-22-TS, Cyclo-23-TS and Proparg-21-TS datasets in different atom-mapping regimes. We show that, compared to existing models for reaction property prediction, 3DReact offers a flexible framework that exploits atom-mapping information, if available, as well as geometries of reactants and products (in an invariant or equivariant fashion). Accordingly, it performs systematically well across different datasets, atom-mapping regimes, as well as both interpolation and extrapolation tasks.
Read more7/15/2024
0
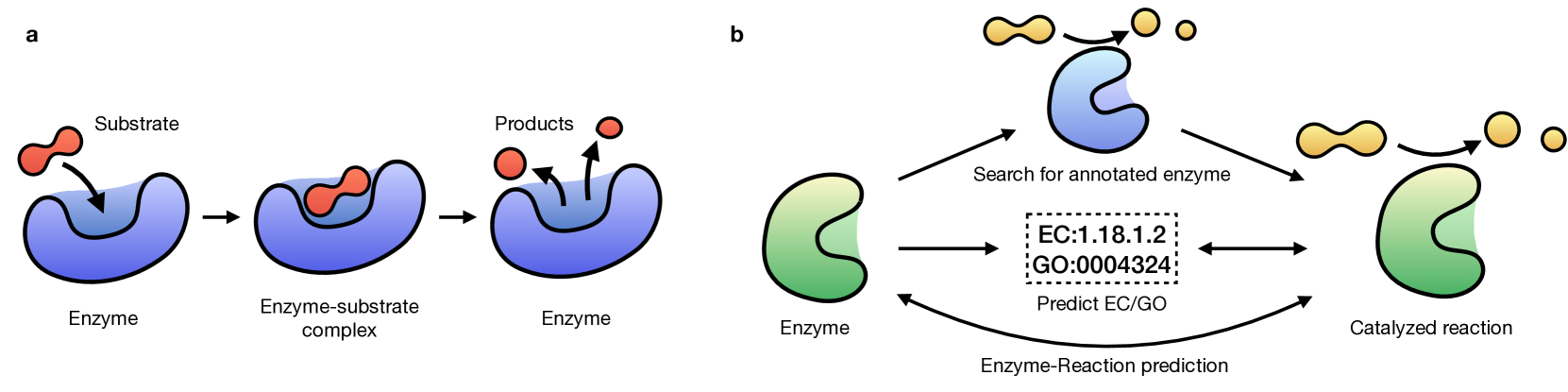
Reactzyme: A Benchmark for Enzyme-Reaction Prediction
Chenqing Hua, Bozitao Zhong, Sitao Luan, Liang Hong, Guy Wolf, Doina Precup, Shuangjia Zheng
Enzymes, with their specific catalyzed reactions, are necessary for all aspects of life, enabling diverse biological processes and adaptations. Predicting enzyme functions is essential for understanding biological pathways, guiding drug development, enhancing bioproduct yields, and facilitating evolutionary studies. Addressing the inherent complexities, we introduce a new approach to annotating enzymes based on their catalyzed reactions. This method provides detailed insights into specific reactions and is adaptable to newly discovered reactions, diverging from traditional classifications by protein family or expert-derived reaction classes. We employ machine learning algorithms to analyze enzyme reaction datasets, delivering a much more refined view on the functionality of enzymes. Our evaluation leverages the largest enzyme-reaction dataset to date, derived from the SwissProt and Rhea databases with entries up to January 8, 2024. We frame the enzyme-reaction prediction as a retrieval problem, aiming to rank enzymes by their catalytic ability for specific reactions. With our model, we can recruit proteins for novel reactions and predict reactions in novel proteins, facilitating enzyme discovery and function annotation.
Read more8/27/2024