Multimodal contrastive learning for spatial gene expression prediction using histology images

0

Sign in to get full access
Overview
- This paper presents a multimodal contrastive learning approach for predicting spatial gene expression from histology images.
- The method learns a joint representation of gene expression and histology image data, allowing it to predict spatial gene expression patterns from histology images alone.
- The authors evaluate their approach on several spatial transcriptomics datasets, demonstrating its ability to accurately predict gene expression distributions.
Plain English Explanation
Genes are the instructions that tell our cells how to function. In the human body, different genes are active in different locations, creating a unique "spatial gene expression" pattern. Understanding these spatial patterns is important for studying disease and development.
However, measuring spatial gene expression directly is complex and expensive. This paper proposes a new way to predict spatial gene expression patterns using commonly available histology images - microscopic pictures of tissue samples. The key idea is to train a machine learning model to learn the relationship between gene expression and histology images.
The model is trained using a "contrastive learning" technique, which learns to map both gene expression data and histology images into a shared representation space. Once trained, the model can take a new histology image as input and predict the corresponding spatial gene expression pattern. This allows researchers to estimate gene expression distributions without the need for specialized spatial transcriptomics experiments.
The authors test their approach on several real-world datasets and show that it can accurately predict spatial gene expression from histology images alone. This could make it easier and more cost-effective to study spatial gene expression patterns in biological research and medical applications.
Technical Explanation
The paper presents a multimodal contrastive learning framework for predicting spatial gene expression from histology images. The core of the approach is a contrastive learning objective that learns a joint representation space for both gene expression data and histology image data.
Specifically, the model takes in paired gene expression and histology image data during training. It learns to map both modalities into a shared latent representation space such that matching gene-image pairs are pushed together, while non-matching pairs are pushed apart. This forces the model to learn features that are predictive of the spatial relationship between gene expression and tissue morphology.
Once trained, the model can take a new histology image as input and use the learned representation to predict the corresponding spatial gene expression pattern. The authors evaluate this capability on several spatial transcriptomics datasets, including STEnTrans, Cross-Modal Diffusion, and STImage-1K4M. They show that their multimodal contrastive approach outperforms previous methods for spatial gene expression prediction from histology images.
Critical Analysis
The paper presents a compelling approach for leveraging readily available histology imaging data to predict spatial gene expression patterns. By learning a joint representation space, the model is able to effectively transfer knowledge from one modality to the other, overcoming the challenge of limited spatial transcriptomics data.
However, the authors note some limitations of their approach. First, the model relies on having paired gene expression and histology data for training, which may not always be available. Additionally, the prediction accuracy is dependent on the quality and representativeness of the training data.
Further research could explore ways to adapt the approach to work with unpaired or noisy data, or to generalize to new tissue types or experimental conditions. There is also potential to combine this method with other techniques, such as super-resolution approaches Cross-Modal Diffusion, to enhance the spatial resolution of the predicted gene expression patterns.
Overall, the work represents an important step forward in the field of spatial genomics, demonstrating how machine learning can be used to extract valuable insights from histology data. As the approach is further developed and applied, it could have significant implications for biological research and clinical applications.
Conclusion
This paper introduces a novel multimodal contrastive learning approach for predicting spatial gene expression patterns from histology images. By learning a joint representation of gene expression and tissue morphology, the model can accurately estimate spatial gene expression distributions without the need for specialized and costly spatial transcriptomics experiments.
The authors demonstrate the effectiveness of their approach on several real-world datasets, outperforming previous methods. While the technique has some limitations, it represents an important advancement in the field of spatial genomics, with the potential to enable more accessible and cost-effective studies of gene expression patterns in biological systems and disease states.
As the method is further refined and combined with other techniques, it could become a valuable tool for researchers and clinicians working to understand the complex relationships between genes, tissue structure, and physiological function.
This summary was produced with help from an AI and may contain inaccuracies - check out the links to read the original source documents!
Related Papers
0
Multimodal contrastive learning for spatial gene expression prediction using histology images
Wenwen Min, Zhiceng Shi, Jun Zhang, Jun Wan, Changmiao Wang
In recent years, the advent of spatial transcriptomics (ST) technology has unlocked unprecedented opportunities for delving into the complexities of gene expression patterns within intricate biological systems. Despite its transformative potential, the prohibitive cost of ST technology remains a significant barrier to its widespread adoption in large-scale studies. An alternative, more cost-effective strategy involves employing artificial intelligence to predict gene expression levels using readily accessible whole-slide images (WSIs) stained with Hematoxylin and Eosin (H&E). However, existing methods have yet to fully capitalize on multimodal information provided by H&E images and ST data with spatial location. In this paper, we propose textbf{mclSTExp}, a multimodal contrastive learning with Transformer and Densenet-121 encoder for Spatial Transcriptomics Expression prediction. We conceptualize each spot as a word, integrating its intrinsic features with spatial context through the self-attention mechanism of a Transformer encoder. This integration is further enriched by incorporating image features via contrastive learning, thereby enhancing the predictive capability of our model. Our extensive evaluation of textbf{mclSTExp} on two breast cancer datasets and a skin squamous cell carcinoma dataset demonstrates its superior performance in predicting spatial gene expression. Moreover, mclSTExp has shown promise in interpreting cancer-specific overexpressed genes, elucidating immune-related genes, and identifying specialized spatial domains annotated by pathologists. Our source code is available at https://github.com/shizhiceng/mclSTExp.
Read more7/12/2024
0
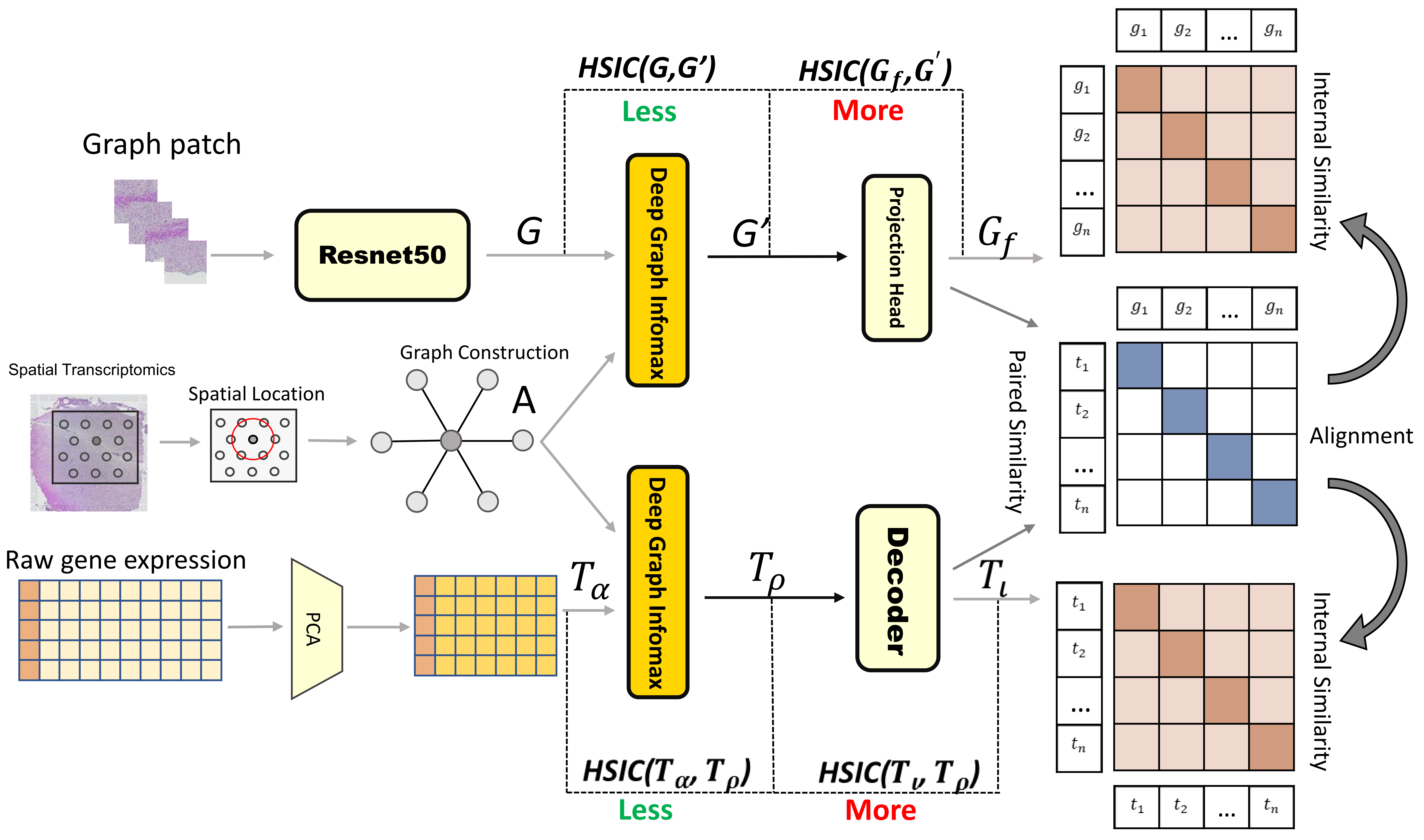
Spatially Resolved Gene Expression Prediction from Histology via Multi-view Graph Contrastive Learning with HSIC-bottleneck Regularization
Changxi Chi, Hang Shi, Qi Zhu, Daoqiang Zhang, Wei Shao
The rapid development of spatial transcriptomics(ST) enables the measurement of gene expression at spatial resolution, making it possible to simultaneously profile the gene expression, spatial locations of spots, and the matched histopathological images. However, the cost for collecting ST data is much higher than acquiring histopathological images, and thus several studies attempt to predict the gene expression on ST by leveraging their corresponding histopathological images. Most of the existing image-based gene prediction models treat the prediction task on each spot of ST data independently, which ignores the spatial dependency among spots. In addition, while the histology images share phenotypic characteristics with the ST data, it is still challenge to extract such common information to help align paired image and expression representations. To address the above issues, we propose a Multi-view Graph Contrastive Learning framework with HSIC-bottleneck Regularization(ST-GCHB) aiming at learning shared representation to help impute the gene expression of the queried imagingspots by considering their spatial dependency.
Read more6/19/2024

0
HistoSPACE: Histology-Inspired Spatial Transcriptome Prediction And Characterization Engine
Shivam Kumar, Samrat Chatterjee
Spatial transcriptomics (ST) enables the visualization of gene expression within the context of tissue morphology. This emerging discipline has the potential to serve as a foundation for developing tools to design precision medicines. However, due to the higher costs and expertise required for such experiments, its translation into a regular clinical practice might be challenging. Despite the implementation of modern deep learning to enhance information obtained from histological images using AI, efforts have been constrained by limitations in the diversity of information. In this paper, we developed a model, HistoSPACE that explore the diversity of histological images available with ST data to extract molecular insights from tissue image. Our proposed study built an image encoder derived from universal image autoencoder. This image encoder was connected to convolution blocks to built the final model. It was further fine tuned with the help of ST-Data. This model is notably lightweight in compared to traditional histological models. Our developed model demonstrates significant efficiency compared to contemporary algorithms, revealing a correlation of 0.56 in leave-one-out cross-validation. Finally, its robustness was validated through an independent dataset, showing a well matched preditction with predefined disease pathology.
Read more8/9/2024

0
stEnTrans: Transformer-based deep learning for spatial transcriptomics enhancement
Shuailin Xue, Fangfang Zhu, Changmiao Wang, Wenwen Min
The spatial location of cells within tissues and organs is crucial for the manifestation of their specific functions.Spatial transcriptomics technology enables comprehensive measurement of the gene expression patterns in tissues while retaining spatial information. However, current popular spatial transcriptomics techniques either have shallow sequencing depth or low resolution. We present stEnTrans, a deep learning method based on Transformer architecture that provides comprehensive predictions for gene expression in unmeasured areas or unexpectedly lost areas and enhances gene expression in original and inputed spots. Utilizing a self-supervised learning approach, stEnTrans establishes proxy tasks on gene expression profile without requiring additional data, mining intrinsic features of the tissues as supervisory information. We evaluate stEnTrans on six datasets and the results indicate superior performance in enhancing spots resolution and predicting gene expression in unmeasured areas compared to other deep learning and traditional interpolation methods. Additionally, Our method also can help the discovery of spatial patterns in Spatial Transcriptomics and enrich to more biologically significant pathways. Our source code is available at https://github.com/shuailinxue/stEnTrans.
Read more7/12/2024