Protein binding affinity prediction under multiple substitutions applying eGNNs on Residue and Atomic graphs combined with Language model information: eGRAL

0
Sign in to get full access
Overview
- Proposes a novel method called eGRAL (e3-equivariant Graph Neural Networks on Residue and Atomic graphs combined with Language model information) for predicting protein binding affinity under multiple substitutions
- Leverages both residue-level and atomic-level graph representations, as well as language model embeddings, to improve prediction performance
- Demonstrates the effectiveness of eGRAL on several protein-protein interaction datasets
Plain English Explanation
Predicting how well two proteins will bind to each other is an important problem in biology and drug discovery. This paper introduces a new method called eGRAL that aims to improve upon existing techniques for predicting protein binding affinity, especially when there are multiple changes (substitutions) to the proteins.
The key idea behind eGRAL is to combine different ways of representing the protein structure into a single model. Specifically, it uses graph neural networks to capture both the residue-level (amino acid) and atomic-level structure of the proteins. It also incorporates information from language models that have been trained on a large amount of protein sequence data.
By integrating these diverse sources of information, eGRAL is able to make more accurate predictions of how strongly two proteins will bind together, even when one or both of the proteins have been modified. This can be useful for designing new protein-based drugs or understanding how mutations affect protein interactions.
Technical Explanation
The eGRAL method consists of three main components:
-
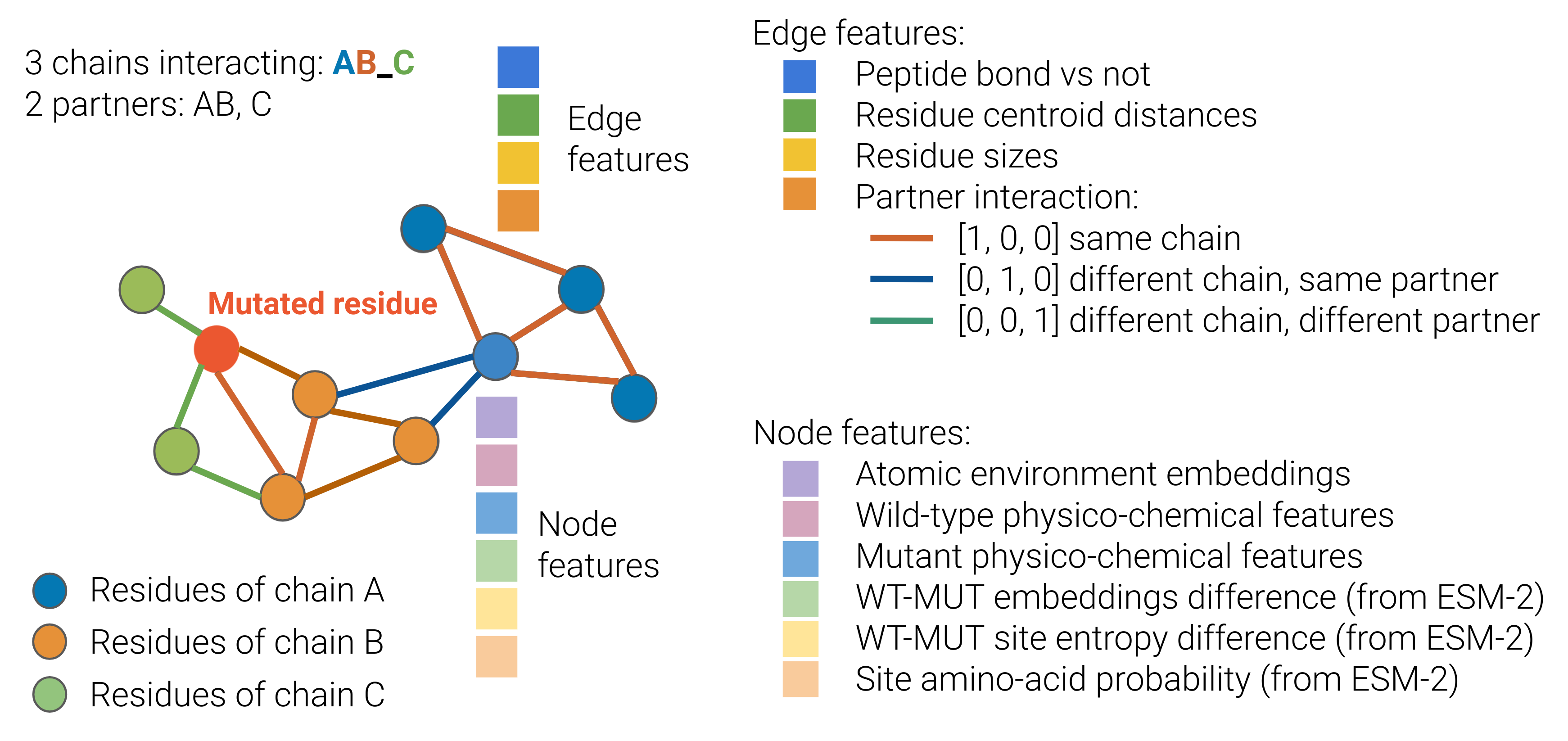
Residue Graph Encoder: This module takes the sequence of amino acids in a protein and constructs a graph representation, where each node represents a residue and edges capture the spatial relationships between residues. e3-equivariant graph neural networks are used to learn features from this residue-level graph.
-
Atomic Graph Encoder: In parallel, eGRAL also constructs a graph representing the atomic structure of the protein, where each node is an individual atom. Another set of e3-equivariant graph neural networks is used to extract features from this atomic-level graph.
-
Language Model Integration: To further enhance the representation, eGRAL incorporates contextual embeddings from a pre-trained language model that has been trained on a large corpus of protein sequence data. These embeddings capture additional information about the semantic and structural properties of the proteins.
The outputs of these three components are then combined and passed through a final neural network to produce the predicted binding affinity between the two input proteins. The authors show that eGRAL outperforms previous state-of-the-art methods for protein binding affinity prediction, especially on datasets with multiple substitutions.
Critical Analysis
The eGRAL paper makes a compelling case for the benefits of integrating multiple representations of protein structure and sequence information to improve binding affinity prediction. The authors provide thorough experimental validation on several benchmark datasets, demonstrating the effectiveness of their approach.
However, the paper does not address certain limitations or potential issues with the method. For example, the computational complexity of constructing and processing the residue-level and atomic-level graphs may be a concern, especially for large protein complexes. Additionally, the reliance on pre-trained language models introduces a dependency on the quality and coverage of the training data used for those models.
Further research could explore ways to make the eGRAL architecture more efficient or investigate alternative approaches for incorporating sequence-based information, such as using transformer-based models or deep reinforcement learning. Comparisons to other graph-based or geometric deep learning methods for protein structure analysis would also be valuable.
Conclusion
The eGRAL method proposed in this paper represents a promising advance in the field of protein binding affinity prediction. By combining residue-level and atomic-level graph representations with language model information, eGRAL demonstrates improved performance, particularly on datasets with multiple protein substitutions. This work highlights the potential benefits of integrating diverse sources of structural and sequence-based data to tackle complex problems in computational biology and drug discovery.
This summary was produced with help from an AI and may contain inaccuracies - check out the links to read the original source documents!
Related Papers
0
Protein binding affinity prediction under multiple substitutions applying eGNNs on Residue and Atomic graphs combined with Language model information: eGRAL
Arturo Fiorellini-Bernardis, Sebastien Boyer, Christoph Brunken, Bakary Diallo, Karim Beguir, Nicolas Lopez-Carranza, Oliver Bent
Protein-protein interactions (PPIs) play a crucial role in numerous biological processes. Developing methods that predict binding affinity changes under substitution mutations is fundamental for modelling and re-engineering biological systems. Deep learning is increasingly recognized as a powerful tool capable of bridging the gap between in-silico predictions and in-vitro observations. With this contribution, we propose eGRAL, a novel SE(3) equivariant graph neural network (eGNN) architecture designed for predicting binding affinity changes from multiple amino acid substitutions in protein complexes. eGRAL leverages residue, atomic and evolutionary scales, thanks to features extracted from protein large language models. To address the limited availability of large-scale affinity assays with structural information, we generate a simulated dataset comprising approximately 500,000 data points. Our model is pre-trained on this dataset, then fine-tuned and tested on experimental data.
Read more5/7/2024
🧠
0
VN-EGNN: E(3)-Equivariant Graph Neural Networks with Virtual Nodes Enhance Protein Binding Site Identification
Florian Sestak, Lisa Schneckenreiter, Johannes Brandstetter, Sepp Hochreiter, Andreas Mayr, Gunter Klambauer
Being able to identify regions within or around proteins, to which ligands can potentially bind, is an essential step to develop new drugs. Binding site identification methods can now profit from the availability of large amounts of 3D structures in protein structure databases or from AlphaFold predictions. Current binding site identification methods heavily rely on graph neural networks (GNNs), usually designed to output E(3)-equivariant predictions. Such methods turned out to be very beneficial for physics-related tasks like binding energy or motion trajectory prediction. However, the performance of GNNs at binding site identification is still limited potentially due to the lack of dedicated nodes that model hidden geometric entities, such as binding pockets. In this work, we extend E(n)-Equivariant Graph Neural Networks (EGNNs) by adding virtual nodes and applying an extended message passing scheme. The virtual nodes in these graphs are dedicated quantities to learn representations of binding sites, which leads to improved predictive performance. In our experiments, we show that our proposed method VN-EGNN sets a new state-of-the-art at locating binding site centers on COACH420, HOLO4K and PDBbind2020.
Read more4/11/2024
💬
0
Ranking protein-protein models with large language models and graph neural networks
Xiaotong Xu, Alexandre M. J. J. Bonvin
Protein-protein interactions (PPIs) are associated with various diseases, including cancer, infections, and neurodegenerative disorders. Obtaining three-dimensional structural information on these PPIs serves as a foundation to interfere with those or to guide drug design. Various strategies can be followed to model those complexes, all typically resulting in a large number of models. A challenging step in this process is the identification of good models (near-native PPI conformations) from the large pool of generated models. To address this challenge, we previously developed DeepRank-GNN-esm, a graph-based deep learning algorithm for ranking modelled PPI structures harnessing the power of protein language models. Here, we detail the use of our software with examples. DeepRank-GNN-esm is freely available at https://github.com/haddocking/DeepRank-GNN-esm
Read more7/24/2024

0
Advanced atom-level representations for protein flexibility prediction utilizing graph neural networks
Sina Sarparast, Aldo Zaimi, Maximilian Ebert, Michael-Rock Goldsmith
Protein dynamics play a crucial role in many biological processes and drug interactions. However, measuring, and simulating protein dynamics is challenging and time-consuming. While machine learning holds promise in deciphering the determinants of protein dynamics from structural information, most existing methods for protein representation learning operate at the residue level, ignoring the finer details of atomic interactions. In this work, we propose for the first time to use graph neural networks (GNNs) to learn protein representations at the atomic level and predict B-factors from protein 3D structures. The B-factor reflects the atomic displacement of atoms in proteins, and can serve as a surrogate for protein flexibility. We compared different GNN architectures to assess their performance. The Meta-GNN model achieves a correlation coefficient of 0.71 on a large and diverse test set of over 4k proteins (17M atoms) from the Protein Data Bank (PDB), outperforming previous methods by a large margin. Our work demonstrates the potential of representations learned by GNNs for protein flexibility prediction and other related tasks.
Read more8/23/2024