Deep Reinforcement Learning for Modelling Protein Complexes

0
🤿
Sign in to get full access
Overview
- This paper proposes a novel approach called Generative Adversarial Policy Network (GAPN) for predicting the structure of multi-chain protein complexes.
- The key challenges addressed are the huge combinatorial optimization space and the need for generalization to complexes of varied scales.
- GAPN uses an acyclic undirected connected graph to represent multi-chain protein complexes and learns to efficiently search the assembly space through policy gradient optimization.
- The model also employs an adversarial reward function to enhance its ability to capture the global assembly rules across complexes with different chain numbers.
Plain English Explanation
Proteins are complex molecules that play crucial roles in our bodies. They can come in different shapes and sizes, and sometimes they work together in groups called protein complexes. Predicting the structure of these protein complexes is an important but very challenging task in biology and drug discovery.
The paper presents a new method called GAPN that can predict the structure of protein complexes with multiple chains (subunits). The authors use an approach where they represent each chain as a node and the connections between them as edges, forming an acyclic graph. This allows them to efficiently search through the vast number of possible ways the chains can assemble.
To address the challenges of this problem, GAPN uses two key techniques. First, it learns to navigate the huge search space by optimizing a direct docking reward through a technique called policy gradient. Second, it employs an adversarial reward function to help the model better understand the general rules governing how protein complexes are assembled, regardless of the specific number of chains involved.
The researchers show that GAPN can predict the structures of multi-chain protein complexes more accurately and much faster than the previous state-of-the-art method. This is an important advance that could benefit fields like molecular modeling and drug design.
Technical Explanation
The paper proposes a Generative Adversarial Policy Network (GAPN) for protein complex modeling (PCM), which aims to predict the 3D structure of multi-chain protein complexes. The authors represent each protein chain as a node in an acyclic undirected connected graph, and the assembly actions as edges, allowing them to formulate PCM as a graph optimization problem.
To address the key challenges of the PCM problem, namely the huge combinatorial optimization space and the need for generalization across complexes of varied scales, GAPN employs two main techniques:
-
Policy Gradient Optimization: GAPN learns to efficiently search the immense assembly space and optimize a direct docking reward through policy gradient, a reinforcement learning technique.
-
Adversarial Reward Function: The authors design an adversarial reward function to enhance the model's ability to capture the global assembly rules learned from complexes with varied chain numbers. This helps GAPN focus on both specific batches of complexes and the overall assembly patterns.
Experimentally, GAPN outperforms the previous state-of-the-art method, MoLPC, by up to 27% in terms of TM-Score (a metric for structural similarity) while being 600 times faster. This demonstrates the effectiveness of GAPN's approaches in predicting the structures of multi-chain protein complexes accurately and efficiently.
Critical Analysis
The paper presents a compelling approach to the challenging problem of protein complex modeling. However, there are a few potential limitations and areas for further research:
-
Generalization to Larger Complexes: While GAPN shows impressive performance on the tested datasets, it remains to be seen how well the model will scale to predict the structures of significantly larger and more complex protein assemblies, which are common in biological systems.
-
Handling Flexibility and Dynamics: The current formulation of GAPN assumes rigid protein chains, but many protein complexes exhibit flexibility and dynamic behavior. Extending the model to capture these aspects could further improve its real-world applicability.
-
Interpretability and Explainability: As with many deep learning models, the inner workings of GAPN may be difficult to interpret. Developing techniques to make the model's decision-making process more transparent could enhance its usability and trust.
-
Integrating Additional Biological Knowledge: The authors primarily leverage the structural information of protein chains. Incorporating additional biological knowledge, such as evolutionary conservation, binding affinities, or experimental data, could potentially further improve the model's performance and reliability.
Overall, GAPN represents an important step forward in the field of protein complex modeling and could have significant implications for various applications in biology and drug discovery. Addressing the identified limitations and exploring further research directions could lead to even more powerful and versatile tools for predicting and understanding the structures of complex biomolecular assemblies.
Conclusion
The paper presents a novel Generative Adversarial Policy Network (GAPN) for efficient and accurate prediction of the 3D structures of multi-chain protein complexes. By representing the complexes as acyclic graphs and leveraging policy gradient optimization and adversarial training, GAPN addresses the key challenges of the protein complex modeling problem. The model's significant improvements in both accuracy and computational efficiency over the previous state-of-the-art demonstrate its potential to advance the field of structural biology and contribute to applications in areas such as drug discovery and molecular modeling. Further research to address the identified limitations and explore additional avenues could lead to even more powerful and versatile tools for understanding the complex world of biomolecular structures.
This summary was produced with help from an AI and may contain inaccuracies - check out the links to read the original source documents!
Related Papers
🤿
0
Deep Reinforcement Learning for Modelling Protein Complexes
Ziqi Gao, Tao Feng, Jiaxuan You, Chenyi Zi, Yan Zhou, Chen Zhang, Jia Li
AlphaFold can be used for both single-chain and multi-chain protein structure prediction, while the latter becomes extremely challenging as the number of chains increases. In this work, by taking each chain as a node and assembly actions as edges, we show that an acyclic undirected connected graph can be used to predict the structure of multi-chain protein complexes (a.k.a., protein complex modelling, PCM). However, there are still two challenges: 1) The huge combinatorial optimization space of $N^{N-2}$ ($N$ is the number of chains) for the PCM problem can easily lead to high computational cost. 2) The scales of protein complexes exhibit distribution shift due to variance in chain numbers, which calls for the generalization in modelling complexes of various scales. To address these challenges, we propose GAPN, a Generative Adversarial Policy Network powered by domain-specific rewards and adversarial loss through policy gradient for automatic PCM prediction. Specifically, GAPN learns to efficiently search through the immense assembly space and optimize the direct docking reward through policy gradient. Importantly, we design an adversarial reward function to enhance the receptive field of our model. In this way, GAPN will simultaneously focus on a specific batch of complexes and the global assembly rules learned from complexes with varied chain numbers. Empirically, we have achieved both significant accuracy (measured by RMSD and TM-Score) and efficiency improvements compared to leading PCM softwares.
Read more5/8/2024

0
Progressive Multi-Modality Learning for Inverse Protein Folding
Jiangbin Zheng, Stan Z. Li
While deep generative models show promise for learning inverse protein folding directly from data, the lack of publicly available structure-sequence pairings limits their generalization. Previous improvements and data augmentation efforts to overcome this bottleneck have been insufficient. To further address this challenge, we propose a novel protein design paradigm called MMDesign, which leverages multi-modality transfer learning. To our knowledge, MMDesign is the first framework that combines a pretrained structural module with a pretrained contextual module, using an auto-encoder (AE) based language model to incorporate prior protein semantic knowledge. Experimental results, only training with the small dataset, demonstrate that MMDesign consistently outperforms baselines on various public benchmarks. To further assess the biological plausibility, we present systematic quantitative analysis techniques that provide interpretability and reveal more about the laws of protein design.
Read more7/23/2024

0
One-step Structure Prediction and Screening for Protein-Ligand Complexes using Multi-Task Geometric Deep Learning
Kelei He, Tiejun Dong, Jinhui Wu, Junfeng Zhang
Understanding the structure of the protein-ligand complex is crucial to drug development. Existing virtual structure measurement and screening methods are dominated by docking and its derived methods combined with deep learning. However, the sampling and scoring methodology have largely restricted the accuracy and efficiency. Here, we show that these two fundamental tasks can be accurately tackled with a single model, namely LigPose, based on multi-task geometric deep learning. By representing the ligand and the protein pair as a graph, LigPose directly optimizes the three-dimensional structure of the complex, with the learning of binding strength and atomic interactions as auxiliary tasks, enabling its one-step prediction ability without docking tools. Extensive experiments show LigPose achieved state-of-the-art performance on major tasks in drug research. Its considerable improvements indicate a promising paradigm of AI-based pipeline for drug development.
Read more8/22/2024
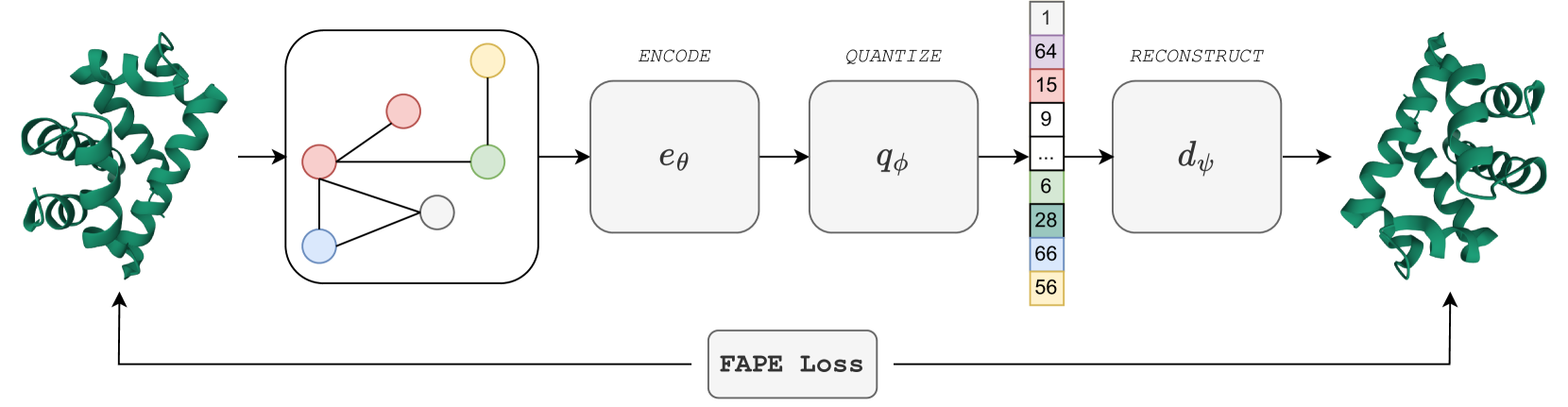
0
Learning the Language of Protein Structure
Benoit Gaujac, J'er'emie Don`a, Liviu Copoiu, Timothy Atkinson, Thomas Pierrot, Thomas D. Barrett
Representation learning and emph{de novo} generation of proteins are pivotal computational biology tasks. Whilst natural language processing (NLP) techniques have proven highly effective for protein sequence modelling, structure modelling presents a complex challenge, primarily due to its continuous and three-dimensional nature. Motivated by this discrepancy, we introduce an approach using a vector-quantized autoencoder that effectively tokenizes protein structures into discrete representations. This method transforms the continuous, complex space of protein structures into a manageable, discrete format with a codebook ranging from 4096 to 64000 tokens, achieving high-fidelity reconstructions with backbone root mean square deviations (RMSD) of approximately 1-5 AA. To demonstrate the efficacy of our learned representations, we show that a simple GPT model trained on our codebooks can generate novel, diverse, and designable protein structures. Our approach not only provides representations of protein structure, but also mitigates the challenges of disparate modal representations and sets a foundation for seamless, multi-modal integration, enhancing the capabilities of computational methods in protein design.
Read more5/28/2024