Gene Regulatory Network Inference from Pre-trained Single-Cell Transcriptomics Transformer with Joint Graph Learning

0

Sign in to get full access
Overview
- Introduces a new approach to infer gene regulatory networks (GRNs) from single-cell RNA sequencing (scRNA-seq) data using a pre-trained transformer model and joint graph learning.
- The proposed method, called !link TransGNN, leverages the power of transformers to capture complex gene expression patterns and jointly learns the GRN structure.
- Demonstrates improved performance on benchmark datasets compared to existing GRN inference methods.
Plain English Explanation
!link Single-cell RNA sequencing (scRNA-seq) is a powerful technique that allows researchers to study gene expression patterns in individual cells. By analyzing these patterns, scientists can infer the underlying !link gene regulatory networks (GRNs) - the complex web of interactions between genes that control cellular processes.
The researchers in this paper developed a new method, called TransGNN, to infer GRNs from scRNA-seq data. TransGNN uses a pre-trained !link transformer model to capture the complex relationships between genes, and then jointly learns the structure of the GRN.
The key idea is that the transformer model can identify subtle patterns in gene expression that traditional methods might miss. By combining this powerful feature extraction with the ability to directly learn the GRN structure, TransGNN can infer more accurate and informative GRNs than previous approaches.
The researchers tested TransGNN on several benchmark datasets and found that it outperformed other state-of-the-art GRN inference methods. This suggests that TransGNN could be a valuable tool for researchers studying the underlying gene regulatory mechanisms in biological systems.
Technical Explanation
The !link TransGNN model consists of two main components: a pre-trained transformer module and a joint graph learning module.
The transformer module is based on the !link Transformer architecture, which has been shown to be effective at capturing complex patterns in sequential data, such as gene expression profiles. The researchers fine-tune this pre-trained transformer on the scRNA-seq data to extract rich features that encode the relationships between genes.
The joint graph learning module then takes these transformer-derived features and learns the structure of the GRN. This is done by optimizing a joint objective function that combines the transformer's learning of gene expression patterns with the direct learning of the GRN adjacency matrix.
By optimizing these two components together, TransGNN is able to leverage the strengths of both the transformer and the graph learning to infer more accurate GRNs compared to previous methods that treat these steps separately.
The researchers evaluate TransGNN on several benchmark scRNA-seq datasets and show that it outperforms state-of-the-art GRN inference algorithms in terms of accuracy, as measured by standard metrics like AUPR and AUROC.
Critical Analysis
The paper presents a compelling approach to GRN inference that leverages the power of transformer models and joint graph learning. The results on benchmark datasets are promising and suggest that TransGNN could be a valuable tool for researchers studying gene regulation.
However, the paper does not address some potential limitations and areas for further research. For example, the performance of TransGNN may be sensitive to the choice of pre-trained transformer model and hyperparameters, which could limit its generalizability. Additionally, the paper does not explore the interpretability of the learned GRNs, which is an important consideration for biological applications.
Further research could investigate ways to improve the robustness and interpretability of TransGNN, such as by incorporating additional biological priors or developing explainable AI techniques. It would also be valuable to test the method on a wider range of scRNA-seq datasets, including those with different characteristics and experimental designs.
Overall, the TransGNN approach represents an exciting advancement in the field of GRN inference, and the paper lays the groundwork for further development and exploration of transformer-based methods in this domain.
Conclusion
The !link paper introduces a novel method called TransGNN for inferring gene regulatory networks from single-cell RNA sequencing data. By leveraging the power of pre-trained transformer models and joint graph learning, TransGNN is able to capture complex gene expression patterns and infer more accurate GRNs compared to existing approaches.
The results demonstrate the potential of this approach to advance our understanding of the underlying gene regulatory mechanisms that govern cellular processes. While the paper highlights some promising directions, further research is needed to address the limitations and expand the applicability of TransGNN. Nonetheless, this work represents an important step forward in the field of computational biology and single-cell genomics.
This summary was produced with help from an AI and may contain inaccuracies - check out the links to read the original source documents!
Related Papers
0
Gene Regulatory Network Inference from Pre-trained Single-Cell Transcriptomics Transformer with Joint Graph Learning
Sindhura Kommu, Yizhi Wang, Yue Wang, Xuan Wang
Inferring gene regulatory networks (GRNs) from single-cell RNA sequencing (scRNA-seq) data is a complex challenge that requires capturing the intricate relationships between genes and their regulatory interactions. In this study, we tackle this challenge by leveraging the single-cell BERT-based pre-trained transformer model (scBERT), trained on extensive unlabeled scRNA-seq data, to augment structured biological knowledge from existing GRNs. We introduce a novel joint graph learning approach that combines the rich contextual representations learned by pre-trained single-cell language models with the structured knowledge encoded in GRNs using graph neural networks (GNNs). By integrating these two modalities, our approach effectively reasons over boththe gene expression level constraints provided by the scRNA-seq data and the structured biological knowledge inherent in GRNs. We evaluate our method on human cell benchmark datasets from the BEELINE study with cell type-specific ground truth networks. The results demonstrate superior performance over current state-of-the-art baselines, offering a deeper understanding of cellular regulatory mechanisms.
Read more7/26/2024
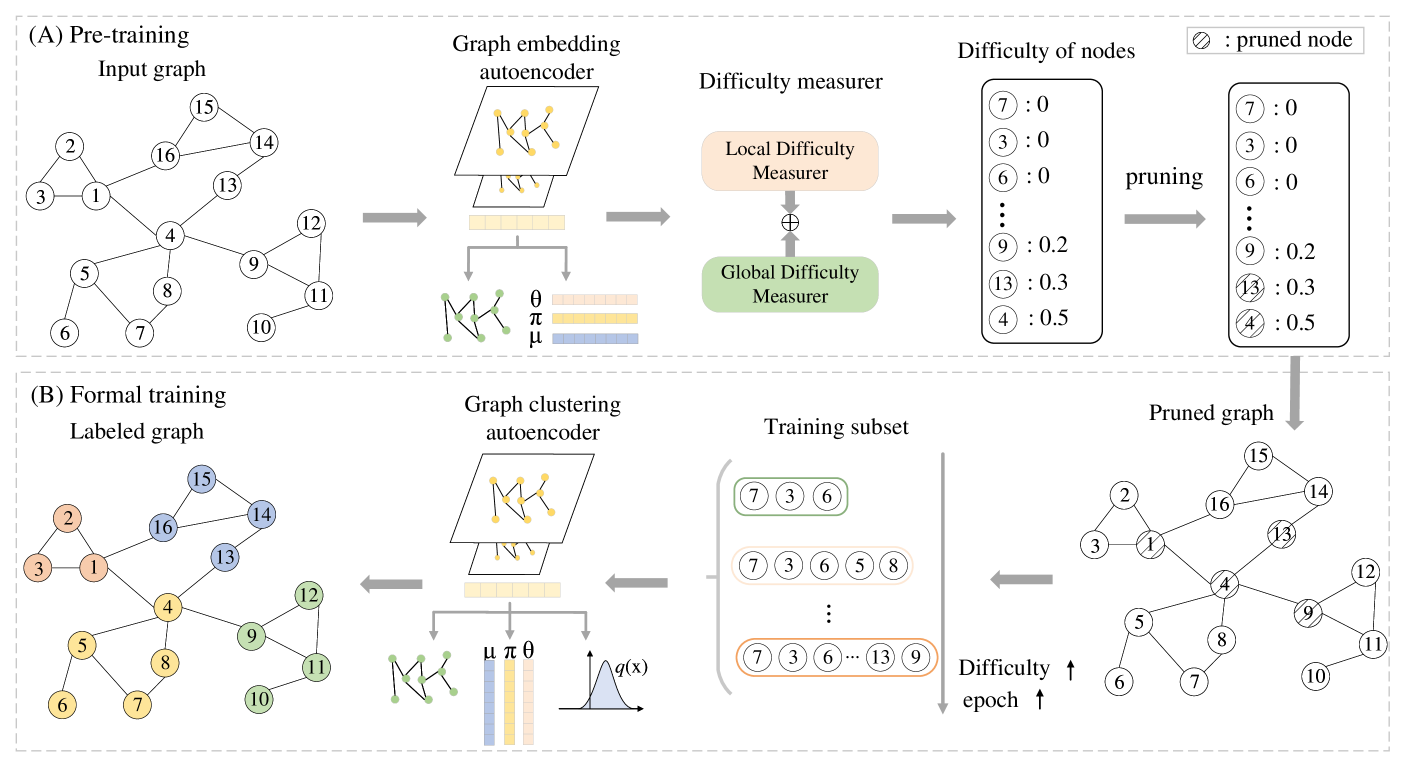
0
Single-cell Curriculum Learning-based Deep Graph Embedding Clustering
Huifa Li, Jie Fu, Xinpeng Ling, Zhiyu Sun, Kuncan Wang, Zhili Chen
The swift advancement of single-cell RNA sequencing (scRNA-seq) technologies enables the investigation of cellular-level tissue heterogeneity. Cell annotation significantly contributes to the extensive downstream analysis of scRNA-seq data. However, The analysis of scRNA-seq for biological inference presents challenges owing to its intricate and indeterminate data distribution, characterized by a substantial volume and a high frequency of dropout events. Furthermore, the quality of training samples varies greatly, and the performance of the popular scRNA-seq data clustering solution GNN could be harmed by two types of low-quality training nodes: 1) nodes on the boundary; 2) nodes that contribute little additional information to the graph. To address these problems, we propose a single-cell curriculum learning-based deep graph embedding clustering (scCLG). We first propose a Chebyshev graph convolutional autoencoder with multi-decoder (ChebAE) that combines three optimization objectives corresponding to three decoders, including topology reconstruction loss of cell graphs, zero-inflated negative binomial (ZINB) loss, and clustering loss, to learn cell-cell topology representation. Meanwhile, we employ a selective training strategy to train GNN based on the features and entropy of nodes and prune the difficult nodes based on the difficulty scores to keep the high-quality graph. Empirical results on a variety of gene expression datasets show that our model outperforms state-of-the-art methods.
Read more8/21/2024
🤿
0
Pan-cancer gene set discovery via scRNA-seq for optimal deep learning based downstream tasks
Jong Hyun Kim, Jongseong Jang
The application of machine learning to transcriptomics data has led to significant advances in cancer research. However, the high dimensionality and complexity of RNA sequencing (RNA-seq) data pose significant challenges in pan-cancer studies. This study hypothesizes that gene sets derived from single-cell RNA sequencing (scRNA-seq) data will outperform those selected using bulk RNA-seq in pan-cancer downstream tasks. We analyzed scRNA-seq data from 181 tumor biopsies across 13 cancer types. High-dimensional weighted gene co-expression network analysis (hdWGCNA) was performed to identify relevant gene sets, which were further refined using XGBoost for feature selection. These gene sets were applied to downstream tasks using TCGA pan-cancer RNA-seq data and compared to six reference gene sets and oncogenes from OncoKB evaluated with deep learning models, including multilayer perceptrons (MLPs) and graph neural networks (GNNs). The XGBoost-refined hdWGCNA gene set demonstrated higher performance in most tasks, including tumor mutation burden assessment, microsatellite instability classification, mutation prediction, cancer subtyping, and grading. In particular, genes such as DPM1, BAD, and FKBP4 emerged as important pan-cancer biomarkers, with DPM1 consistently significant across tasks. This study presents a robust approach for feature selection in cancer genomics by integrating scRNA-seq data and advanced analysis techniques, offering a promising avenue for improving predictive accuracy in cancer research.
Read more8/15/2024

0
scCDCG: Efficient Deep Structural Clustering for single-cell RNA-seq via Deep Cut-informed Graph Embedding
Ping Xu, Zhiyuan Ning, Meng Xiao, Guihai Feng, Xin Li, Yuanchun Zhou, Pengfei Wang
Single-cell RNA sequencing (scRNA-seq) is essential for unraveling cellular heterogeneity and diversity, offering invaluable insights for bioinformatics advancements. Despite its potential, traditional clustering methods in scRNA-seq data analysis often neglect the structural information embedded in gene expression profiles, crucial for understanding cellular correlations and dependencies. Existing strategies, including graph neural networks, face challenges in handling the inefficiency due to scRNA-seq data's intrinsic high-dimension and high-sparsity. Addressing these limitations, we introduce scCDCG (single-cell RNA-seq Clustering via Deep Cut-informed Graph), a novel framework designed for efficient and accurate clustering of scRNA-seq data that simultaneously utilizes intercellular high-order structural information. scCDCG comprises three main components: (i) A graph embedding module utilizing deep cut-informed techniques, which effectively captures intercellular high-order structural information, overcoming the over-smoothing and inefficiency issues prevalent in prior graph neural network methods. (ii) A self-supervised learning module guided by optimal transport, tailored to accommodate the unique complexities of scRNA-seq data, specifically its high-dimension and high-sparsity. (iii) An autoencoder-based feature learning module that simplifies model complexity through effective dimension reduction and feature extraction. Our extensive experiments on 6 datasets demonstrate scCDCG's superior performance and efficiency compared to 7 established models, underscoring scCDCG's potential as a transformative tool in scRNA-seq data analysis. Our code is available at: https://github.com/XPgogogo/scCDCG.
Read more4/10/2024