Autoregressive Enzyme Function Prediction with Multi-scale Multi-modality Fusion

0
Sign in to get full access
Overview
- This paper proposes a novel autoregressive enzyme function prediction model that leverages multi-scale multi-modality fusion.
- The model aims to accurately predict enzyme functions by integrating various types of data, including protein sequences, structures, and annotations.
- The researchers conducted experiments to evaluate the performance of their approach on several benchmark datasets.
Plain English Explanation
The paper describes a new machine learning model for predicting the functions of enzymes. Enzymes are proteins that speed up chemical reactions in living organisms. Understanding an enzyme's function is important for many applications, such as designing new enzymes or developing drugs.
The researchers developed a model that can take in different types of data about an enzyme, including its amino acid sequence and 3D structure, and use that information to predict what the enzyme's function is. Their approach is unique in that it combines these various data sources at different "scales" or levels of detail to get a more complete understanding of the enzyme.
By testing their model on established benchmark datasets, the researchers showed that it can make more accurate function predictions compared to previous methods. This suggests their approach could be a valuable tool for advancing our knowledge of enzymes and their roles in biological systems.
Technical Explanation
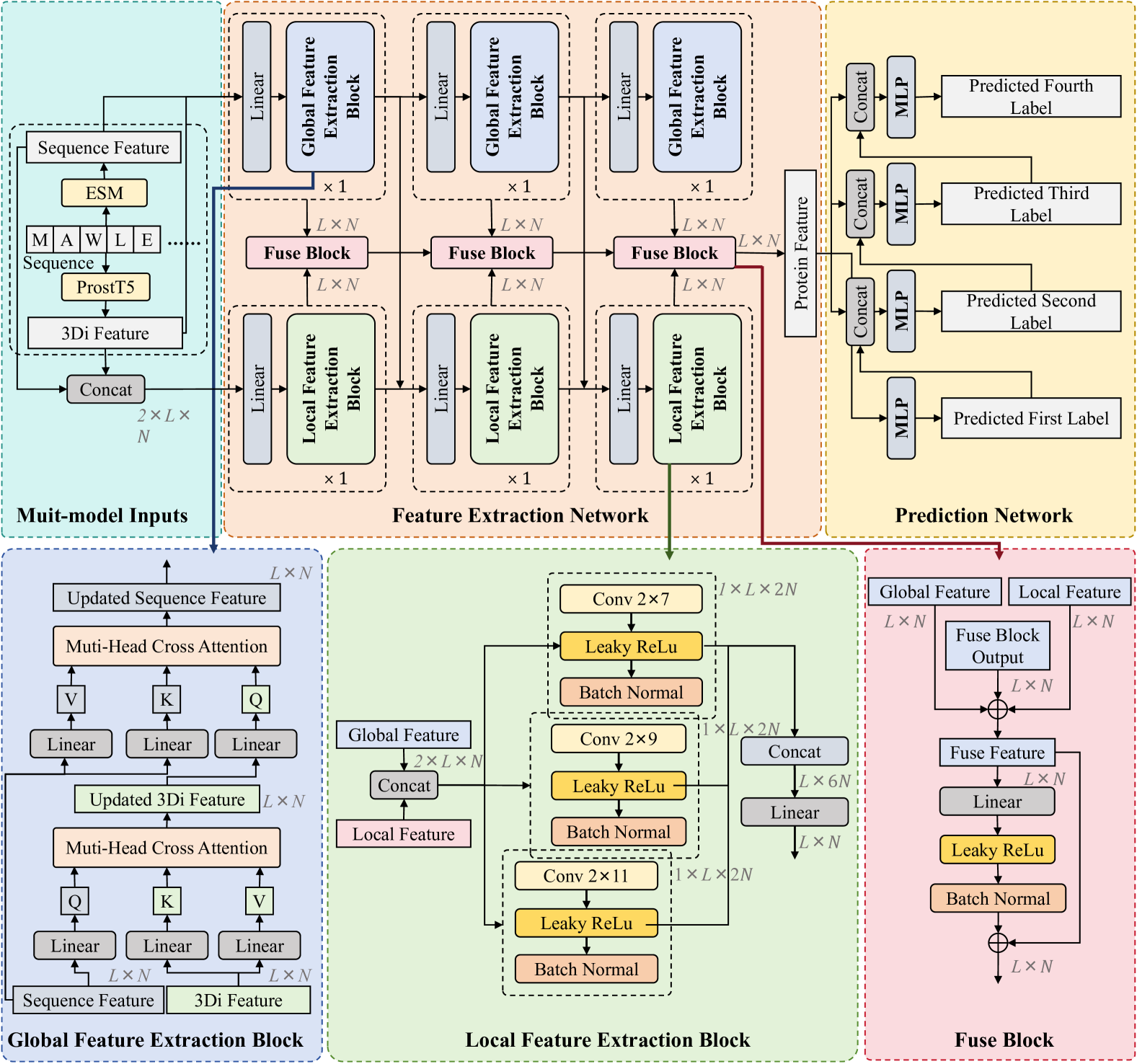
The key innovation of this paper is the development of an autoregressive enzyme function prediction model that fuses multi-scale multi-modality data. The model takes in various types of enzyme data, including protein sequences, structures, and annotations, and learns to predict the enzyme's function in an iterative, step-by-step manner.
The architecture of the model consists of several components. First, it uses transformer-based encoders to extract features from the input data at different scales, such as the amino acid level, secondary structure level, and whole-protein level. These multi-scale representations are then combined using attention mechanisms to capture interdependencies between the different data modalities.
The fused multi-scale features are fed into an autoregressive decoder that generates the enzyme function prediction one step at a time. This allows the model to iteratively refine its understanding of the enzyme's function based on the previously predicted outputs.
The researchers evaluated their model on several standard benchmark datasets for enzyme function prediction, including UniProtKB/Swiss-Prot and ProtFun. They showed that their approach outperforms previous state-of-the-art methods in terms of prediction accuracy, demonstrating the benefits of their multi-scale multi-modality fusion strategy.
Critical Analysis
The paper makes a strong methodological contribution by introducing a novel autoregressive architecture that effectively integrates diverse enzyme data sources. The multi-scale fusion approach is well-motivated and the experimental results are compelling.
However, the paper does not deeply discuss potential limitations or caveats of the proposed method. For example, it is unclear how the model would perform on more challenging or noisy real-world enzyme data, or how robust it is to missing or incomplete input information.
Additionally, while the paper highlights the model's improved predictive performance, it does not provide much insight into the specific biological mechanisms or principles it is leveraging to make those predictions. A more in-depth analysis of the model's internal workings and learned representations could shed light on new enzyme function discovery principles.
Further research is needed to better understand the generalizability and interpretability of this autoregressive multi-scale fusion approach. Exploring its applicability to other domains beyond enzymes, as well as developing techniques to extract biological insights from the model, could be fruitful avenues for future work.
Conclusion
This paper presents a innovative autoregressive enzyme function prediction model that fuses multi-scale multi-modality data to achieve state-of-the-art performance on benchmark tasks. By effectively combining diverse sources of enzyme information, the model can make more accurate function predictions compared to previous methods.
The technical contributions of the paper, particularly the multi-scale fusion architecture, demonstrate the power of leveraging complementary data sources for complex biological inference tasks. While further research is needed to fully understand the model's limitations and potential, this work represents an important step forward in advancing enzyme function prediction capabilities with practical applications in areas like drug discovery and biocatalyst engineering.
This summary was produced with help from an AI and may contain inaccuracies - check out the links to read the original source documents!
Related Papers
0
Autoregressive Enzyme Function Prediction with Multi-scale Multi-modality Fusion
Dingyi Rong, Wenzhuo Zheng, Bozitao Zhong, Zhouhan Lin, Liang Hong, Ning Liu
Accurate prediction of enzyme function is crucial for elucidating biological mechanisms and driving innovation across various sectors. Existing deep learning methods tend to rely solely on either sequence data or structural data and predict the EC number as a whole, neglecting the intrinsic hierarchical structure of EC numbers. To address these limitations, we introduce MAPred, a novel multi-modality and multi-scale model designed to autoregressively predict the EC number of proteins. MAPred integrates both the primary amino acid sequence and the 3D tokens of proteins, employing a dual-pathway approach to capture comprehensive protein characteristics and essential local functional sites. Additionally, MAPred utilizes an autoregressive prediction network to sequentially predict the digits of the EC number, leveraging the hierarchical organization of EC classifications. Evaluations on benchmark datasets, including New-392, Price, and New-815, demonstrate that our method outperforms existing models, marking a significant advance in the reliability and granularity of protein function prediction within bioinformatics.
Read more8/14/2024
0
Reactzyme: A Benchmark for Enzyme-Reaction Prediction
Chenqing Hua, Bozitao Zhong, Sitao Luan, Liang Hong, Guy Wolf, Doina Precup, Shuangjia Zheng
Enzymes, with their specific catalyzed reactions, are necessary for all aspects of life, enabling diverse biological processes and adaptations. Predicting enzyme functions is essential for understanding biological pathways, guiding drug development, enhancing bioproduct yields, and facilitating evolutionary studies. Addressing the inherent complexities, we introduce a new approach to annotating enzymes based on their catalyzed reactions. This method provides detailed insights into specific reactions and is adaptable to newly discovered reactions, diverging from traditional classifications by protein family or expert-derived reaction classes. We employ machine learning algorithms to analyze enzyme reaction datasets, delivering a much more refined view on the functionality of enzymes. Our evaluation leverages the largest enzyme-reaction dataset to date, derived from the SwissProt and Rhea databases with entries up to January 8, 2024. We frame the enzyme-reaction prediction as a retrieval problem, aiming to rank enzymes by their catalytic ability for specific reactions. With our model, we can recruit proteins for novel reactions and predict reactions in novel proteins, facilitating enzyme discovery and function annotation.
Read more8/27/2024
0
Protein Representation Learning by Capturing Protein Sequence-Structure-Function Relationship
Eunji Ko, Seul Lee, Minseon Kim, Dongki Kim
The goal of protein representation learning is to extract knowledge from protein databases that can be applied to various protein-related downstream tasks. Although protein sequence, structure, and function are the three key modalities for a comprehensive understanding of proteins, existing methods for protein representation learning have utilized only one or two of these modalities due to the difficulty of capturing the asymmetric interrelationships between them. To account for this asymmetry, we introduce our novel asymmetric multi-modal masked autoencoder (AMMA). AMMA adopts (1) a unified multi-modal encoder to integrate all three modalities into a unified representation space and (2) asymmetric decoders to ensure that sequence latent features reflect structural and functional information. The experiments demonstrate that the proposed AMMA is highly effective in learning protein representations that exhibit well-aligned inter-modal relationships, which in turn makes it effective for various downstream protein-related tasks.
Read more5/14/2024

0
Generative Enzyme Design Guided by Functionally Important Sites and Small-Molecule Substrates
Zhenqiao Song, Yunlong Zhao, Wenxian Shi, Wengong Jin, Yang Yang, Lei Li
Enzymes are genetically encoded biocatalysts capable of accelerating chemical reactions. How can we automatically design functional enzymes? In this paper, we propose EnzyGen, an approach to learn a unified model to design enzymes across all functional families. Our key idea is to generate an enzyme's amino acid sequence and their three-dimensional (3D) coordinates based on functionally important sites and substrates corresponding to a desired catalytic function. These sites are automatically mined from enzyme databases. EnzyGen consists of a novel interleaving network of attention and neighborhood equivariant layers, which captures both long-range correlation in an entire protein sequence and local influence from nearest amino acids in 3D space. To learn the generative model, we devise a joint training objective, including a sequence generation loss, a position prediction loss and an enzyme-substrate interaction loss. We further construct EnzyBench, a dataset with 3157 enzyme families, covering all available enzymes within the protein data bank (PDB). Experimental results show that our EnzyGen consistently achieves the best performance across all 323 testing families, surpassing the best baseline by 10.79% in terms of substrate binding affinity. These findings demonstrate EnzyGen's superior capability in designing well-folded and effective enzymes binding to specific substrates with high affinities.
Read more7/18/2024