CrysAtom: Distributed Representation of Atoms for Crystal Property Prediction

0
Sign in to get full access
Overview
- Paper proposes a new distributed representation of atoms called CrysAtom for predicting crystal properties
- CrysAtom aims to capture both the chemical and spatial information of atoms in a crystal structure
- The representation is used to predict various crystal properties like formation energy, band gap, and more
Plain English Explanation
The paper introduces a new way to represent atoms in crystal structures, called CrysAtom. Crystals are materials made up of repeating patterns of atoms. To understand and predict the properties of crystals, it's important to capture information about the individual atoms and how they are arranged.
CrysAtom aims to create a more comprehensive representation of atoms that includes both their chemical properties (like the type of element) and their spatial information (like where they are located in the crystal). This combined representation can then be used to predict various crystal properties, such as how much energy is needed to form the crystal, what its electronic band gap is, and more.
The key idea is to learn a distributed representation of each atom, which means encoding the atom's information into a vector of numbers. This allows the model to capture complex relationships between atoms in an efficient way. The authors show that this CrysAtom representation outperforms previous approaches on a range of crystal property prediction tasks.
Technical Explanation
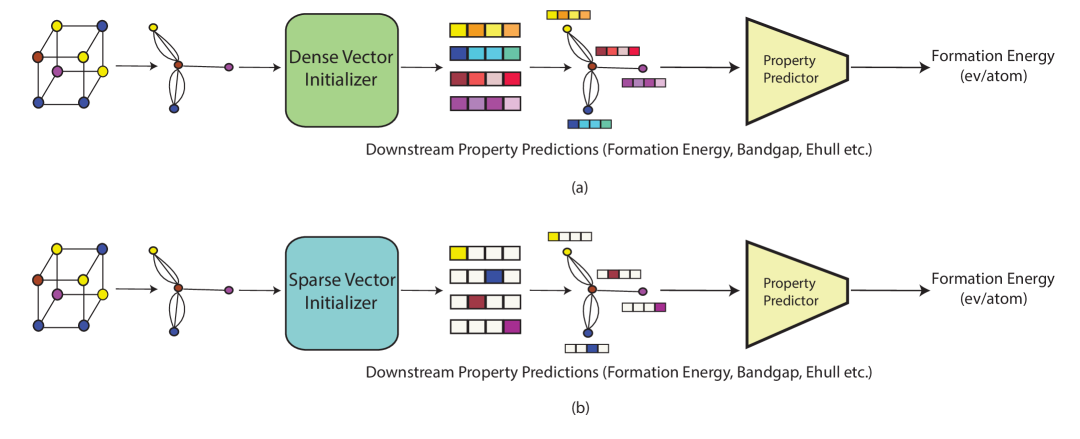
The paper introduces a new atomic representation called CrysAtom for predicting crystal properties. The representation is designed to capture both the chemical and spatial information of atoms in a crystal structure.
The CrysAtom representation is learned using a neural network model that takes as input the crystal's atom types and their 3D coordinates. The model learns a distributed representation for each atom, encoding its properties into a vector of numbers.
To predict a crystal property, the CrysAtom representations of all atoms in the crystal are aggregated using a permutation-invariant function, such as summation or attention. This aggregated representation is then passed through additional neural network layers to predict the target crystal property.
The authors evaluate the CrysAtom representation on several crystal property prediction tasks, including formation energy, band gap, and more. They show that CrysAtom outperforms previous approaches that use other atomic representations, demonstrating the effectiveness of their proposed method.
Critical Analysis
The paper presents a novel and well-designed approach for representing atoms in crystal structures to enable more accurate prediction of crystal properties. The CrysAtom representation captures both chemical and spatial information about atoms, which is important for understanding crystal structures.
One potential limitation is that the CrysAtom representation is learned in a supervised way, requiring labeled data for crystal properties. It would be interesting to see if the representation could also be learned in an unsupervised manner, which may make it more generally applicable.
Additionally, the paper focuses on predicting static crystal properties, but it could be valuable to extend the CrysAtom representation to also capture dynamic behavior, such as how crystals respond to changes in temperature or pressure.
Overall, the CrysAtom approach represents a significant advance in the field of crystal property prediction and could have important implications for materials science and engineering.
Conclusion
This paper introduces a new distributed representation of atoms called CrysAtom that aims to capture both the chemical and spatial information of atoms in a crystal structure. The CrysAtom representation is used to predict various crystal properties, demonstrating its effectiveness compared to previous approaches.
The CrysAtom method represents an important advance in the field of materials informatics, as it provides a more comprehensive way to model the complex relationships between atoms in crystals. This could lead to improved prediction of crystal properties, which has important implications for the design and discovery of new materials with desirable characteristics.
This summary was produced with help from an AI and may contain inaccuracies - check out the links to read the original source documents!
Related Papers
0
CrysAtom: Distributed Representation of Atoms for Crystal Property Prediction
Shrimon Mukherjee, Madhusudan Ghosh, Partha Basuchowdhuri
Application of artificial intelligence (AI) has been ubiquitous in the growth of research in the areas of basic sciences. Frequent use of machine learning (ML) and deep learning (DL) based methodologies by researchers has resulted in significant advancements in the last decade. These techniques led to notable performance enhancements in different tasks such as protein structure prediction, drug-target binding affinity prediction, and molecular property prediction. In material science literature, it is well-known that crystalline materials exhibit topological structures. Such topological structures may be represented as graphs and utilization of graph neural network (GNN) based approaches could help encoding them into an augmented representation space. Primarily, such frameworks adopt supervised learning techniques targeted towards downstream property prediction tasks on the basis of electronic properties (formation energy, bandgap, total energy, etc.) and crystalline structures. Generally, such type of frameworks rely highly on the handcrafted atom feature representations along with the structural representations. In this paper, we propose an unsupervised framework namely, CrysAtom, using untagged crystal data to generate dense vector representation of atoms, which can be utilized in existing GNN-based property predictor models to accurately predict important properties of crystals. Empirical results show that our dense representation embeds chemical properties of atoms and enhance the performance of the baseline property predictor models significantly.
Read more9/10/2024

0
AlphaCrystal-II: Distance matrix based crystal structure prediction using deep learning
Yuqi Song, Rongzhi Dong, Lai Wei, Qin Li, Jianjun Hu
Computational prediction of stable crystal structures has a profound impact on the large-scale discovery of novel functional materials. However, predicting the crystal structure solely from a material's composition or formula is a promising yet challenging task, as traditional ab initio crystal structure prediction (CSP) methods rely on time-consuming global searches and first-principles free energy calculations. Inspired by the recent success of deep learning approaches in protein structure prediction, which utilize pairwise amino acid interactions to describe 3D structures, we present AlphaCrystal-II, a novel knowledge-based solution that exploits the abundant inter-atomic interaction patterns found in existing known crystal structures. AlphaCrystal-II predicts the atomic distance matrix of a target crystal material and employs this matrix to reconstruct its 3D crystal structure. By leveraging the wealth of inter-atomic relationships of known crystal structures, our approach demonstrates remarkable effectiveness and reliability in structure prediction through comprehensive experiments. This work highlights the potential of data-driven methods in accelerating the discovery and design of new materials with tailored properties.
Read more4/9/2024

0
Advanced atom-level representations for protein flexibility prediction utilizing graph neural networks
Sina Sarparast, Aldo Zaimi, Maximilian Ebert, Michael-Rock Goldsmith
Protein dynamics play a crucial role in many biological processes and drug interactions. However, measuring, and simulating protein dynamics is challenging and time-consuming. While machine learning holds promise in deciphering the determinants of protein dynamics from structural information, most existing methods for protein representation learning operate at the residue level, ignoring the finer details of atomic interactions. In this work, we propose for the first time to use graph neural networks (GNNs) to learn protein representations at the atomic level and predict B-factors from protein 3D structures. The B-factor reflects the atomic displacement of atoms in proteins, and can serve as a surrogate for protein flexibility. We compared different GNN architectures to assess their performance. The Meta-GNN model achieves a correlation coefficient of 0.71 on a large and diverse test set of over 4k proteins (17M atoms) from the Protein Data Bank (PDB), outperforming previous methods by a large margin. Our work demonstrates the potential of representations learned by GNNs for protein flexibility prediction and other related tasks.
Read more8/23/2024

0
Predicting Many Properties of Crystals by a Single Deep Learning Model
Haosheng Xu, Dongheng Qian, Jing Wang
The use of machine learning methods for predicting the properties of crystalline materials encounters significant challenges, primarily related to input encoding, output versatility, and interpretability. Here, we introduce CrystalBERT, an adaptable transformer-based framework with novel structure that integrates space group, elemental, and unit cell information. The method's adaptability lies not only in its ability to seamlessly combine diverse features but also in its capability to accurately predict a wide range of physically important properties, including topological properties, superconducting transition temperatures, dielectric constants, and more. CrystalBERT also provides insightful physical interpretations regarding the features that most significantly influence the target properties. Our findings indicate that space group and elemental information are more important for predicting topological and superconducting properties, in contrast to some properties that primarily depend on the unit cell information. This underscores the intricate nature of topological and superconducting properties. By incorporating all these features, we achieve a high accuracy of 91% in topological classification, surpassing prior studies and identifying previously misclassified topological materials, further demonstrating the effectiveness of our model.
Read more5/30/2024