EquiPocket: an E(3)-Equivariant Geometric Graph Neural Network for Ligand Binding Site Prediction

0
🧠
Sign in to get full access
Overview
- This paper proposes a new deep learning model called EquiPocket for predicting the binding sites of target proteins, which is a critical task in drug discovery.
- Existing deep learning methods for this problem often treat proteins as 3D images and use 3D convolutional neural networks (CNNs), but these models have several limitations.
- EquiPocket uses an E(3)-equivariant Graph Neural Network (GNN) to address the shortcomings of CNN-based approaches.
Plain English Explanation
Predicting where drugs or other molecules will bind to proteins is an important step in developing new medicines. Most current AI models for this task treat the protein as a 3D image and use 3D convolutional neural networks (CNNs) to analyze it. However, these CNN-based methods have some key problems:
- They struggle to represent the irregular, complex shapes of proteins.
- They are sensitive to how the protein is rotated or oriented.
- They don't capture the full details of the protein's surface very well.
- They don't adapt well when the size of the protein changes.
To address these limitations, the researchers developed a new AI model called EquiPocket. Instead of a 3D image, EquiPocket uses a graph neural network to represent the protein. This allows it to better handle the irregular structure and geometry of proteins.
EquiPocket has three key components:
- One module extracts detailed information about the local geometry around each atom on the protein's surface.
- Another module models both the chemical and spatial structure of the entire protein.
- The final module uses "equivariant message passing" to capture the overall shape and geometry of the protein's surface.
EquiPocket also includes a "dense attention" output layer to help it handle proteins of different sizes.
The researchers tested EquiPocket on several standard benchmarks and found it outperformed other state-of-the-art methods for predicting protein binding sites. This suggests EquiPocket could be a valuable tool to aid drug discovery efforts.
Technical Explanation
The key innovation in EquiPocket is its use of an E(3)-equivariant Graph Neural Network (GNN) to model the protein structure. Unlike CNN-based approaches, which treat the protein as a 3D image, EquiPocket represents the protein as a graph with atoms as nodes and chemical bonds as edges.
EquiPocket has three main modules:
-
Local Geometry Extraction: This module extracts detailed geometric information about the local environment around each surface atom of the protein. This includes properties like the positions, types, and orientations of neighboring atoms.
-
Protein Structure Modeling: The second module takes the local geometric features and models the overall chemical and spatial structure of the full protein using an E(3)-equivariant GNN. This allows EquiPocket to capture the irregular shapes and symmetries inherent to protein structures.
-
Surface Geometry Encoding: The final module uses equivariant message passing over the protein surface atoms to encode the global geometric features of the protein's surface. This helps EquiPocket characterize the binding site locations more effectively.
Additionally, EquiPocket employs a "dense attention" output layer to address the problem of variable protein sizes. This allows the model to adaptively focus on the most relevant parts of the protein when making predictions.
The researchers evaluated EquiPocket on several standard benchmarks for protein-ligand binding site prediction, including the PDBbind and Binding MOAD datasets. EquiPocket demonstrated superior performance compared to other state-of-the-art deep learning methods, suggesting its potential to advance the field of computational drug discovery.
Critical Analysis
The authors acknowledge several limitations of their work that warrant further research:
-
Computational Efficiency: While EquiPocket outperforms other models, it may have higher computational requirements due to the complexity of the E(3)-equivariant GNN architecture. Optimizing the model's efficiency will be important for real-world applications.
-
Generalization Ability: The authors only evaluated EquiPocket on a limited number of benchmark datasets. More extensive testing is needed to assess how well the model generalizes to a broader range of protein structures and binding site prediction tasks.
-
Interpretability: As with many deep learning models, the internal workings of EquiPocket can be difficult to interpret. Developing more explainable AI techniques for protein binding site prediction could help researchers understand the model's decision-making process and gain deeper insights.
-
Integrating Domain Knowledge: The authors note that EquiPocket could potentially be improved by incorporating additional domain-specific knowledge about protein structures and binding mechanisms. Exploring ways to seamlessly integrate such information into the model architecture could be a fruitful area for future research.
Overall, the EquiPocket model represents a promising advancement in the field of computational drug discovery, but continued research and development will be necessary to address its current limitations and further enhance its capabilities.
Conclusion
This paper presents EquiPocket, a novel deep learning model for predicting the binding sites of target proteins, which is a crucial task in drug discovery. EquiPocket addresses several shortcomings of existing CNN-based approaches by using an E(3)-equivariant Graph Neural Network to better capture the irregular geometry and symmetries inherent to protein structures.
The key innovations in EquiPocket include its ability to extract detailed local geometric features, model the overall chemical and spatial structure of proteins, and encode the global surface geometry using equivariant message passing. Additionally, the model's dense attention output layer helps it handle proteins of varying sizes.
Experimental results on standard benchmarks show that EquiPocket outperforms other state-of-the-art methods for protein binding site prediction, suggesting its potential to advance computational drug discovery. However, the authors also identify several areas for future work, such as improving the model's computational efficiency, exploring its generalization abilities, and enhancing its interpretability.
Overall, the EquiPocket model represents an important step forward in the development of AI tools to assist in the complex and crucial task of drug discovery.
This summary was produced with help from an AI and may contain inaccuracies - check out the links to read the original source documents!
Related Papers
🧠
0
EquiPocket: an E(3)-Equivariant Geometric Graph Neural Network for Ligand Binding Site Prediction
Yang Zhang, Zhewei Wei, Ye Yuan, Chongxuan Li, Wenbing Huang
Predicting the binding sites of target proteins plays a fundamental role in drug discovery. Most existing deep-learning methods consider a protein as a 3D image by spatially clustering its atoms into voxels and then feed the voxelized protein into a 3D CNN for prediction. However, the CNN-based methods encounter several critical issues: 1) defective in representing irregular protein structures; 2) sensitive to rotations; 3) insufficient to characterize the protein surface; 4) unaware of protein size shift. To address the above issues, this work proposes EquiPocket, an E(3)-equivariant Graph Neural Network (GNN) for binding site prediction, which comprises three modules: the first one to extract local geometric information for each surface atom, the second one to model both the chemical and spatial structure of protein and the last one to capture the geometry of the surface via equivariant message passing over the surface atoms. We further propose a dense attention output layer to alleviate the effect incurred by variable protein size. Extensive experiments on several representative benchmarks demonstrate the superiority of our framework to the state-of-the-art methods.
Read more7/24/2024
🧠
0
VN-EGNN: E(3)-Equivariant Graph Neural Networks with Virtual Nodes Enhance Protein Binding Site Identification
Florian Sestak, Lisa Schneckenreiter, Johannes Brandstetter, Sepp Hochreiter, Andreas Mayr, Gunter Klambauer
Being able to identify regions within or around proteins, to which ligands can potentially bind, is an essential step to develop new drugs. Binding site identification methods can now profit from the availability of large amounts of 3D structures in protein structure databases or from AlphaFold predictions. Current binding site identification methods heavily rely on graph neural networks (GNNs), usually designed to output E(3)-equivariant predictions. Such methods turned out to be very beneficial for physics-related tasks like binding energy or motion trajectory prediction. However, the performance of GNNs at binding site identification is still limited potentially due to the lack of dedicated nodes that model hidden geometric entities, such as binding pockets. In this work, we extend E(n)-Equivariant Graph Neural Networks (EGNNs) by adding virtual nodes and applying an extended message passing scheme. The virtual nodes in these graphs are dedicated quantities to learn representations of binding sites, which leads to improved predictive performance. In our experiments, we show that our proposed method VN-EGNN sets a new state-of-the-art at locating binding site centers on COACH420, HOLO4K and PDBbind2020.
Read more4/11/2024
🧠
0
A hybrid quantum-classical fusion neural network to improve protein-ligand binding affinity predictions for drug discovery
L. Domingo, M. Chehimi, S. Banerjee, S. He Yuxun, S. Konakanchi, L. Ogunfowora, S. Roy, S. Selvaras, M. Djukic, C. Johnson
The field of drug discovery hinges on the accurate prediction of binding affinity between prospective drug molecules and target proteins, especially when such proteins directly influence disease progression. However, estimating binding affinity demands significant financial and computational resources. While state-of-the-art methodologies employ classical machine learning (ML) techniques, emerging hybrid quantum machine learning (QML) models have shown promise for enhanced performance, owing to their inherent parallelism and capacity to manage exponential increases in data dimensionality. Despite these advances, existing models encounter issues related to convergence stability and prediction accuracy. This paper introduces a novel hybrid quantum-classical deep learning model tailored for binding affinity prediction in drug discovery. Specifically, the proposed model synergistically integrates 3D and spatial graph convolutional neural networks within an optimized quantum architecture. Simulation results demonstrate a 6% improvement in prediction accuracy relative to existing classical models, as well as a significantly more stable convergence performance compared to previous classical approaches.
Read more9/4/2024
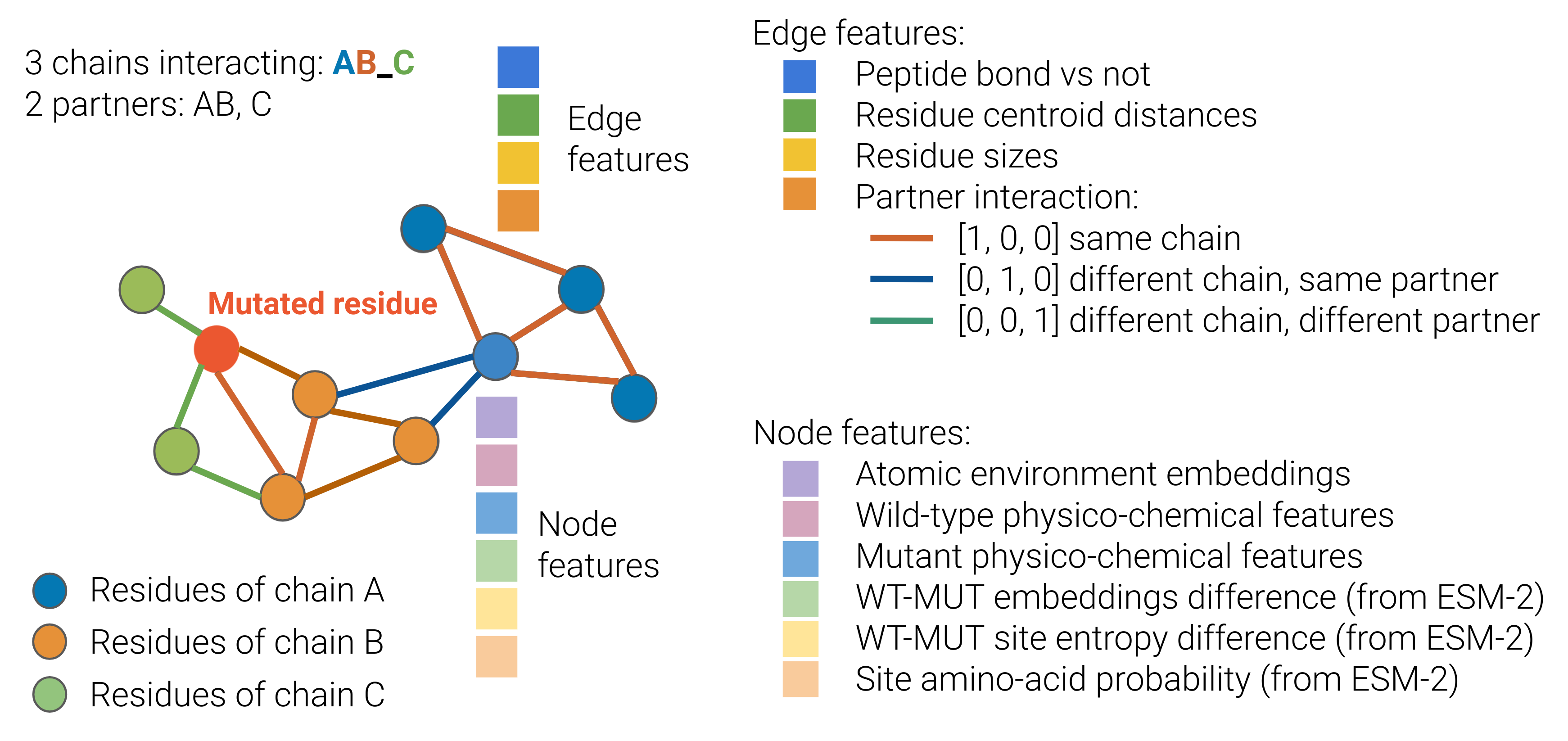
0
Protein binding affinity prediction under multiple substitutions applying eGNNs on Residue and Atomic graphs combined with Language model information: eGRAL
Arturo Fiorellini-Bernardis, Sebastien Boyer, Christoph Brunken, Bakary Diallo, Karim Beguir, Nicolas Lopez-Carranza, Oliver Bent
Protein-protein interactions (PPIs) play a crucial role in numerous biological processes. Developing methods that predict binding affinity changes under substitution mutations is fundamental for modelling and re-engineering biological systems. Deep learning is increasingly recognized as a powerful tool capable of bridging the gap between in-silico predictions and in-vitro observations. With this contribution, we propose eGRAL, a novel SE(3) equivariant graph neural network (eGNN) architecture designed for predicting binding affinity changes from multiple amino acid substitutions in protein complexes. eGRAL leverages residue, atomic and evolutionary scales, thanks to features extracted from protein large language models. To address the limited availability of large-scale affinity assays with structural information, we generate a simulated dataset comprising approximately 500,000 data points. Our model is pre-trained on this dataset, then fine-tuned and tested on experimental data.
Read more5/7/2024