FoldToken2: Learning compact, invariant and generative protein structure language

0
Sign in to get full access
Overview
- This paper presents FoldToken2, a new method for learning a compact, invariant, and generative language for representing protein structures.
- The method leverages recent advances in self-supervised learning and language models to capture the rich information encoded in protein 3D structures.
- FoldToken2 can be used for tasks like protein structure prediction, design, and analysis.
Plain English Explanation
Proteins are the fundamental building blocks of life, carrying out a vast array of essential functions in our bodies. Understanding the 3D structure of proteins is crucial for fields like medicine and biotechnology, as it helps us understand how they work and how we can design new ones.
FoldToken2: Learning compact, invariant and generative protein structure language introduces a new way to represent protein structures using a language model. Just like words in a natural language, the authors have developed a set of "tokens" that can be used to represent the 3D shape of a protein. This allows them to apply powerful machine learning techniques developed for natural language processing to the problem of protein structure analysis.
The key advantage of this approach is that it can learn a very compact and efficient representation of protein structures, which is important for tasks like protein design and simulation. Additionally, the learned representation is invariant to rotations and translations, meaning it can be used to compare and analyze proteins regardless of their orientation.
Technical Explanation
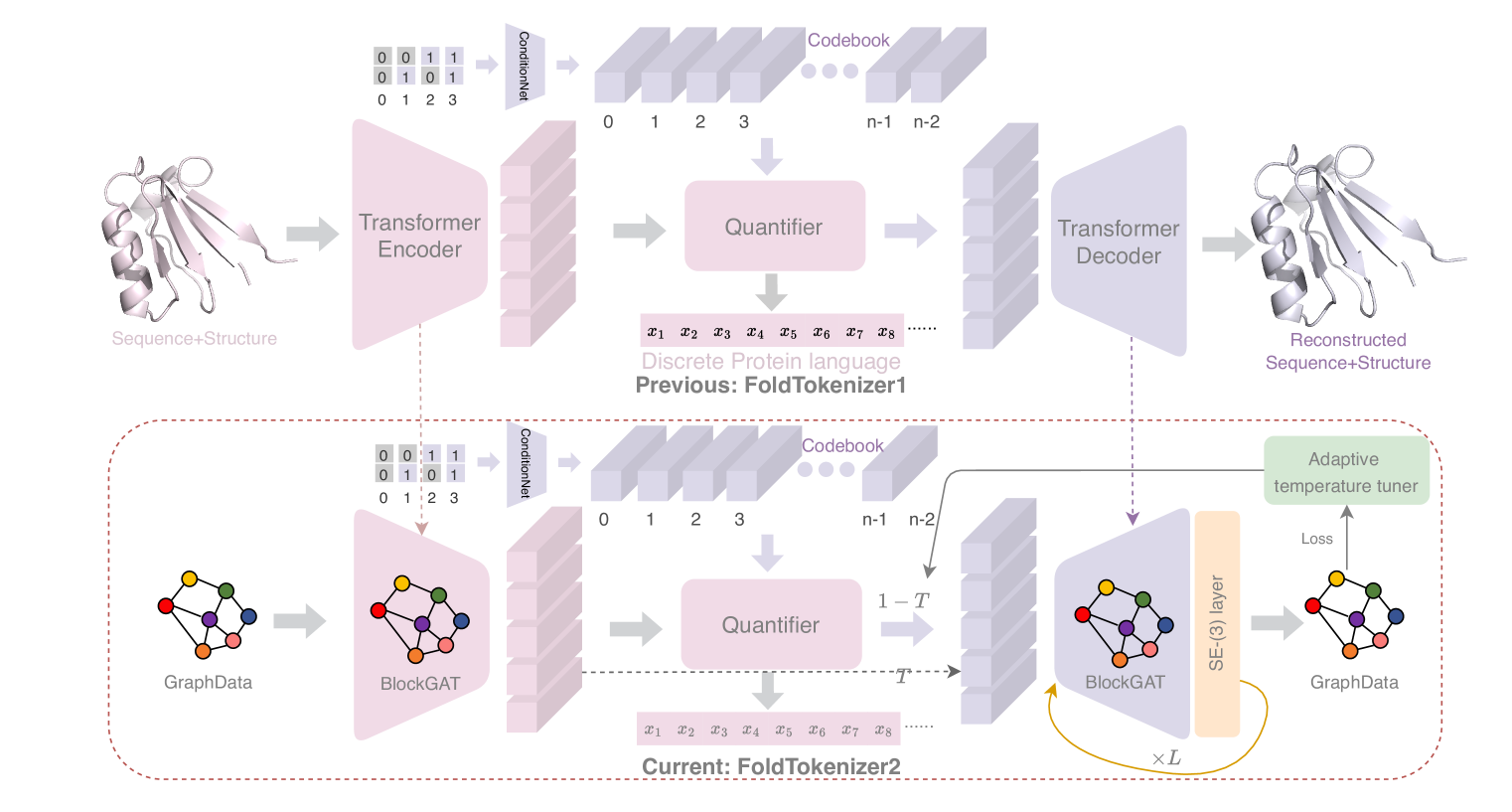
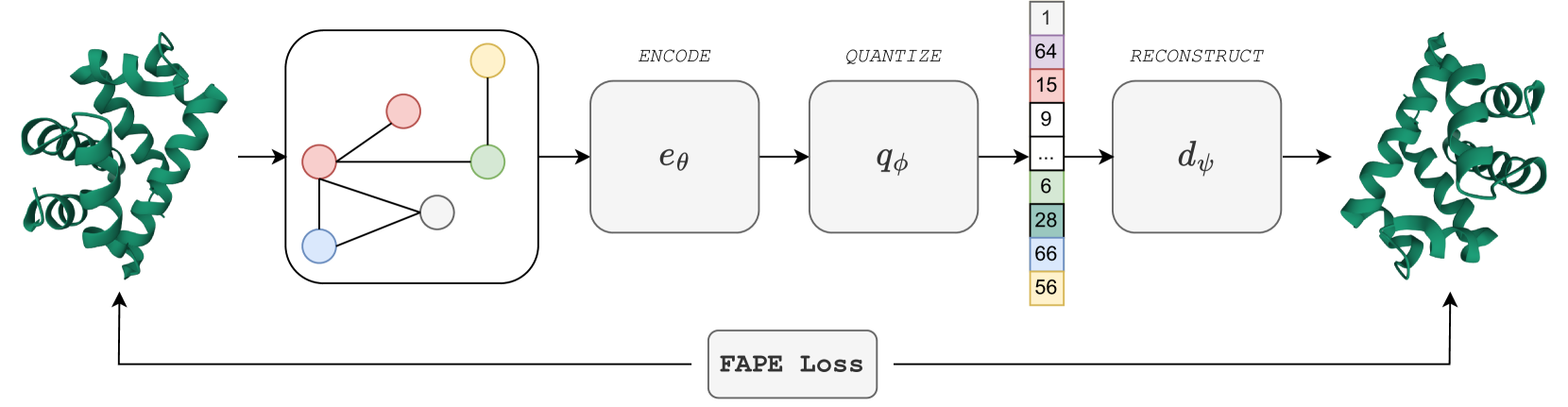
The core idea behind FoldToken2 is to learn a discrete representation of protein structures that captures the essential 3D information in a compact and efficient way. To do this, the authors use a self-supervised learning approach, training a neural network to predict the "tokens" that represent a given protein structure.
The network takes as input the atomic coordinates of a protein and learns to output a sequence of tokens that uniquely identifies that structure. The authors show that this learned representation is highly compact, with just a few hundred tokens sufficient to represent most proteins, yet it retains the key 3D information needed for downstream tasks.
Importantly, the token representations are designed to be invariant to rotations and translations, so that the same protein in different orientations will be represented the same way. This makes the representations useful for tasks like protein structure comparison and alignment.
The authors also demonstrate that the learned token representations can be used to generate new protein structures, by training a generative model to output realistic sequences of tokens. This opens up the possibility of using FoldToken2 for protein design and engineering applications.
Critical Analysis
The FoldToken2 approach represents an interesting and promising new direction for protein structure representation and analysis. The ability to capture the essential 3D information of proteins in a compact, invariant, and generative way is a significant advance over previous methods.
However, the authors acknowledge that the learned token representations are not a perfect, lossless encoding of protein structure. There may be some information lost in the discretization process, which could limit the accuracy of the representations for certain tasks.
Additionally, the generative capabilities of the model are still relatively primitive, and generating completely novel, functional protein structures remains a significant challenge. Further research is needed to improve the generative capabilities and explore the full potential of this approach.
It will also be important to carefully evaluate the representations learned by FoldToken2 on a wide range of protein structure analysis and design tasks, to fully understand its strengths and limitations compared to other state-of-the-art methods, such as Sequence Augmented SE(3) Flow: Matching Conditional Protein Structures and 3D-MolT5: Towards a Unified 3D Molecule Text-to-Structure Model.
Conclusion
In summary, the FoldToken2 method represents an exciting step forward in the representation and analysis of protein structures. By learning a compact, invariant, and generative language for describing protein 3D shapes, the authors have opened up new possibilities for protein design, simulation, and understanding. While further research is needed to fully realize the potential of this approach, it is a promising development that could have significant impacts in fields like medicine, biotechnology, and structural biology.
This summary was produced with help from an AI and may contain inaccuracies - check out the links to read the original source documents!
Related Papers
0
FoldToken2: Learning compact, invariant and generative protein structure language
Zhangyang Gao, Cheng Tan, Stan Z. Li
The equivalent nature of 3D coordinates has posed long term challenges in protein structure representation learning, alignment, and generation. Can we create a compact and invariant language that equivalently represents protein structures? Towards this goal, we propose FoldToken2 to transfer equivariant structures into discrete tokens, while maintaining the recoverability of the original structures. From FoldToken1 to FoldToken2, we improve three key components: (1) invariant structure encoder, (2) vector-quantized compressor, and (3) equivalent structure decoder. We evaluate FoldToken2 on the protein structure reconstruction task and show that it outperforms previous FoldToken1 by 20% in TMScore and 81% in RMSD. FoldToken2 probably be the first method that works well on both single-chain and multi-chain protein structures quantization. We believe that FoldToken2 will inspire further improvement in protein structure representation learning, structure alignment, and structure generation tasks.
Read more7/2/2024
0
Learning the Language of Protein Structure
Benoit Gaujac, J'er'emie Don`a, Liviu Copoiu, Timothy Atkinson, Thomas Pierrot, Thomas D. Barrett
Representation learning and emph{de novo} generation of proteins are pivotal computational biology tasks. Whilst natural language processing (NLP) techniques have proven highly effective for protein sequence modelling, structure modelling presents a complex challenge, primarily due to its continuous and three-dimensional nature. Motivated by this discrepancy, we introduce an approach using a vector-quantized autoencoder that effectively tokenizes protein structures into discrete representations. This method transforms the continuous, complex space of protein structures into a manageable, discrete format with a codebook ranging from 4096 to 64000 tokens, achieving high-fidelity reconstructions with backbone root mean square deviations (RMSD) of approximately 1-5 AA. To demonstrate the efficacy of our learned representations, we show that a simple GPT model trained on our codebooks can generate novel, diverse, and designable protein structures. Our approach not only provides representations of protein structure, but also mitigates the challenges of disparate modal representations and sets a foundation for seamless, multi-modal integration, enhancing the capabilities of computational methods in protein design.
Read more5/28/2024
🤖
0
Synthesizing Proteins on the Graphics Card. Protein Folding and the Limits of Critical AI Studies
Fabian Offert, Paul Kim, Qiaoyu Cai
This paper investigates the application of the transformer architecture in protein folding, as exemplified by DeepMind's AlphaFold project, and its implications for the understanding of large language models as models of language. The prevailing discourse often assumes a ready-made analogy between proteins -- encoded as sequences of amino acids -- and natural language -- encoded as sequences of discrete symbols. Instead of assuming as given the linguistic structure of proteins, we critically evaluate this analogy to assess the kind of knowledge-making afforded by the transformer architecture. We first trace the analogy's emergence and historical development, carving out the influence of structural linguistics on structural biology beginning in the mid-20th century. We then examine three often overlooked pre-processing steps essential to the transformer architecture, including subword tokenization, word embedding, and positional encoding, to demonstrate its regime of representation based on continuous, high-dimensional vector spaces, which departs from the discrete, semantically demarcated symbols of language. The successful deployment of transformers in protein folding, we argue, discloses what we consider a non-linguistic approach to token processing intrinsic to the architecture. We contend that through this non-linguistic processing, the transformer architecture carves out unique epistemological territory and produces a new class of knowledge, distinct from established domains. We contend that our search for intelligent machines has to begin with the shape, rather than the place, of intelligence. Consequently, the emerging field of critical AI studies should take methodological inspiration from the history of science in its quest to conceptualize the contributions of artificial intelligence to knowledge-making, within and beyond the domain-specific sciences.
Read more5/17/2024
0
Evaluating representation learning on the protein structure universe
Arian R. Jamasb, Alex Morehead, Chaitanya K. Joshi, Zuobai Zhang, Kieran Didi, Simon V. Mathis, Charles Harris, Jian Tang, Jianlin Cheng, Pietro Lio, Tom L. Blundell
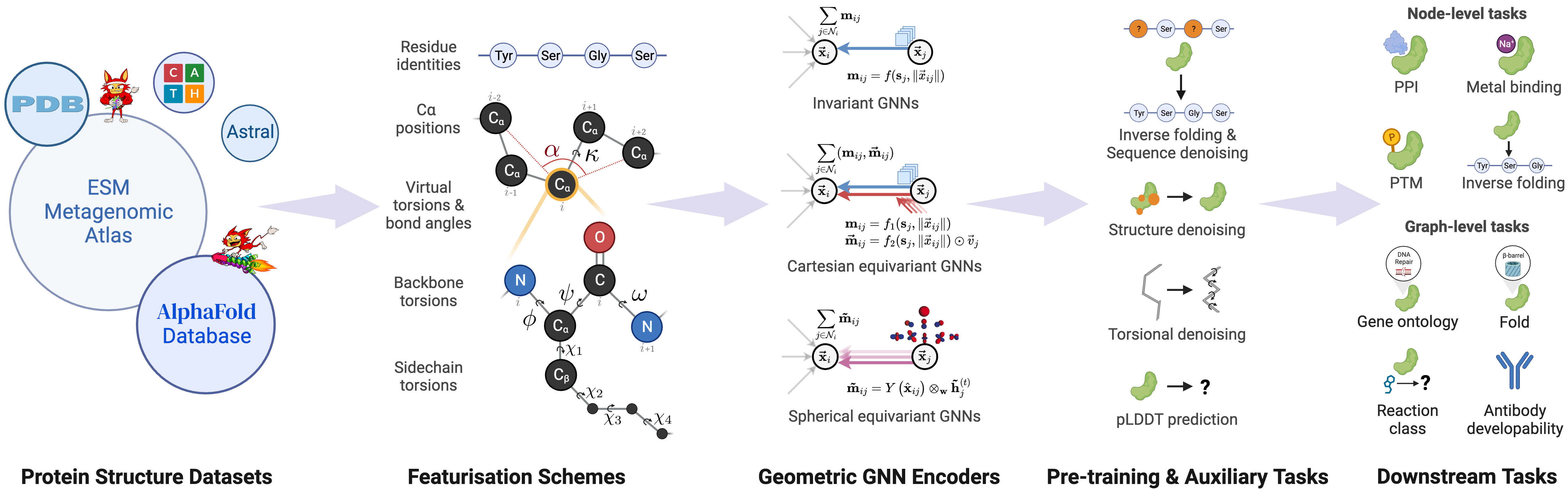
We introduce ProteinWorkshop, a comprehensive benchmark suite for representation learning on protein structures with Geometric Graph Neural Networks. We consider large-scale pre-training and downstream tasks on both experimental and predicted structures to enable the systematic evaluation of the quality of the learned structural representation and their usefulness in capturing functional relationships for downstream tasks. We find that: (1) large-scale pretraining on AlphaFold structures and auxiliary tasks consistently improve the performance of both rotation-invariant and equivariant GNNs, and (2) more expressive equivariant GNNs benefit from pretraining to a greater extent compared to invariant models. We aim to establish a common ground for the machine learning and computational biology communities to rigorously compare and advance protein structure representation learning. Our open-source codebase reduces the barrier to entry for working with large protein structure datasets by providing: (1) storage-efficient dataloaders for large-scale structural databases including AlphaFoldDB and ESM Atlas, as well as (2) utilities for constructing new tasks from the entire PDB. ProteinWorkshop is available at: github.com/a-r-j/ProteinWorkshop.
Read more6/21/2024