From Molecules to Materials: Pre-training Large Generalizable Models for Atomic Property Prediction
2310.16802
0
0
🔮
Abstract
Foundation models have been transformational in machine learning fields such as natural language processing and computer vision. Similar success in atomic property prediction has been limited due to the challenges of training effective models across multiple chemical domains. To address this, we introduce Joint Multi-domain Pre-training (JMP), a supervised pre-training strategy that simultaneously trains on multiple datasets from different chemical domains, treating each dataset as a unique pre-training task within a multi-task framework. Our combined training dataset consists of $sim$120M systems from OC20, OC22, ANI-1x, and Transition-1x. We evaluate performance and generalization by fine-tuning over a diverse set of downstream tasks and datasets including: QM9, rMD17, MatBench, QMOF, SPICE, and MD22. JMP demonstrates an average improvement of 59% over training from scratch, and matches or sets state-of-the-art on 34 out of 40 tasks. Our work highlights the potential of pre-training strategies that utilize diverse data to advance property prediction across chemical domains, especially for low-data tasks. Please visit https://nima.sh/jmp for further information.
Create account to get full access
Overview
- Researchers introduce a new pre-training strategy called Joint Multi-domain Pre-training (JMP) to address the challenges of training effective models across multiple chemical domains.
- JMP simultaneously trains on multiple datasets from different chemical domains, treating each dataset as a unique pre-training task within a multi-task framework.
- The combined training dataset consists of around 120M systems from various sources, including OC20, OC22, ANI-1x, and Transition-1x.
- The researchers evaluate the performance and generalization of JMP by fine-tuning on a diverse set of downstream tasks and datasets, including QM9, rMD17, MatBench, QMOF, SPICE, and MD22.
Plain English Explanation
Predicting the properties of chemicals and materials is an important task in fields like chemistry and materials science. Transformers and other foundation models have revolutionized machine learning in areas like natural language processing and computer vision. However, achieving similar success in predicting atomic properties has been challenging due to the complexity of working with multiple chemical domains.
The researchers developed a new pre-training strategy called Joint Multi-domain Pre-training (JMP) to address this challenge. JMP simultaneously trains the model on multiple datasets from different chemical domains, treating each dataset as a unique pre-training task. This allows the model to learn representations that are applicable across a wide range of chemical systems.
The researchers combined around 120 million chemical systems from various sources, including databases like OC20, OC22, ANI-1x, and Transition-1x, to create the training dataset for JMP. They then evaluated the model's performance and generalization by fine-tuning it on a diverse set of downstream tasks and datasets, such as QM9, rMD17, MatBench, QMOF, SPICE, and MD22.
The results show that JMP can achieve significant improvements over training from scratch, with an average improvement of 59%. Additionally, JMP matches or sets the state-of-the-art on 34 out of 40 tasks, demonstrating its potential to advance property prediction across chemical domains, especially for tasks with limited data.
Technical Explanation
The researchers introduce Joint Multi-domain Pre-training (JMP), a supervised pre-training strategy that simultaneously trains on multiple datasets from different chemical domains. This approach treats each dataset as a unique pre-training task within a multi-task framework, allowing the model to learn representations that are applicable across a wide range of chemical systems.
The combined training dataset used in this study consists of approximately 120 million systems from various sources, including OC20, OC22, ANI-1x, and Transition-1x. The researchers evaluate the performance and generalization of JMP by fine-tuning the pre-trained model on a diverse set of downstream tasks and datasets, including QM9, rMD17, MatBench, QMOF, SPICE, and MD22.
The results demonstrate that JMP achieves an average improvement of 59% over training from scratch, and matches or sets the state-of-the-art on 34 out of 40 tasks. This highlights the potential of pre-training strategies that leverage diverse data to advance property prediction across chemical domains, particularly for tasks with limited available data.
The role of model architecture and scale in predicting molecular properties is an important consideration in this research, as the choice of model and the amount of training data can significantly impact the model's performance.
Critical Analysis
The researchers provide a thorough evaluation of JMP's performance across a diverse set of downstream tasks and datasets, which is a strength of the study. However, the paper could have provided more details on the specific architectures and hyperparameters used, as well as the computational resources required to train the models, to allow for better reproducibility and understanding of the technical aspects.
Additionally, the paper does not address potential biases or limitations in the training datasets, which could impact the model's generalization to real-world scenarios. Further research could explore the robustness of JMP to out-of-distribution samples or its performance on more challenging or realistic chemical prediction tasks.
Learning quantum properties from short-range correlations is another relevant area of research that could provide insights into the types of chemical features and representations that are most important for accurate property prediction.
Conclusion
The Joint Multi-domain Pre-training (JMP) approach introduced in this paper demonstrates the potential of leveraging diverse data and multi-task learning to advance property prediction across chemical domains. By simultaneously training on multiple datasets, JMP is able to learn representations that generalize well to a wide range of downstream tasks, particularly those with limited available data.
The promising results, with an average improvement of 59% over training from scratch and state-of-the-art performance on 34 out of 40 tasks, highlight the importance of developing effective pre-training strategies for chemical property prediction. This research paves the way for further advancements in the field, with potential applications in areas such as drug discovery, materials design, and environmental chemistry.
This summary was produced with help from an AI and may contain inaccuracies - check out the links to read the original source documents!
Related Papers

Predicting Polymer Properties Based on Multimodal Multitask Pretraining
Fanmeng Wang, Wentao Guo, Minjie Cheng, Shen Yuan, Hongteng Xu, Zhifeng Gao
0
0
In the past few decades, polymers, high-molecular-weight compounds formed by bonding numerous identical or similar monomers covalently, have played an essential role in various scientific fields. In this context, accurate prediction of their properties is becoming increasingly crucial. Typically, the properties of a polymer, such as plasticity, conductivity, bio-compatibility, and so on, are highly correlated with its 3D structure. However, current methods for predicting polymer properties heavily rely on information from polymer SMILES sequences (P-SMILES strings) while ignoring crucial 3D structural information, leading to sub-optimal performance. In this work, we propose MMPolymer, a novel multimodal multitask pretraining framework incorporating both polymer 1D sequential information and 3D structural information to enhance downstream polymer property prediction tasks. Besides, to overcome the limited availability of polymer 3D data, we further propose the Star Substitution strategy to extract 3D structural information effectively. During pretraining, MMPolymer not only predicts masked tokens and recovers 3D coordinates but also achieves the cross-modal alignment of latent representation. Subsequently, we further fine-tune the pretrained MMPolymer for downstream polymer property prediction tasks in the supervised learning paradigm. Experimental results demonstrate that MMPolymer achieves state-of-the-art performance in various polymer property prediction tasks. Moreover, leveraging the pretrained MMPolymer and using only one modality (either P-SMILES string or 3D conformation) during fine-tuning can also surpass existing polymer property prediction methods, highlighting the exceptional capability of MMPolymer in polymer feature extraction and utilization. Our online platform for polymer property prediction is available at https://app.bohrium.dp.tech/mmpolymer.
6/10/2024

New!Impact of Domain Knowledge and Multi-Modality on Intelligent Molecular Property Prediction: A Systematic Survey
Taojie Kuang, Pengfei Liu, Zhixiang Ren
0
0
The precise prediction of molecular properties is essential for advancements in drug development, particularly in virtual screening and compound optimization. The recent introduction of numerous deep learning-based methods has shown remarkable potential in enhancing molecular property prediction (MPP), especially improving accuracy and insights into molecular structures. Yet, two critical questions arise: does the integration of domain knowledge augment the accuracy of molecular property prediction and does employing multi-modal data fusion yield more precise results than unique data source methods? To explore these matters, we comprehensively review and quantitatively analyze recent deep learning methods based on various benchmarks. We discover that integrating molecular information significantly improves molecular property prediction (MPP) for both regression and classification tasks. Specifically, regression improvements, measured by reductions in root mean square error (RMSE), are up to 4.0%, while classification enhancements, measured by the area under the receiver operating characteristic curve (ROC-AUC), are up to 1.7%. We also discover that enriching 2D graphs with 1D SMILES boosts multi-modal learning performance for regression tasks by up to 9.1%, and augmenting 2D graphs with 3D information increases performance for classification tasks by up to 13.2%, with both enhancements measured using ROC-AUC. The two consolidated insights offer crucial guidance for future advancements in drug discovery.
7/1/2024

Transformers for molecular property prediction: Lessons learned from the past five years
Afnan Sultan, Jochen Sieg, Miriam Mathea, Andrea Volkamer
0
0
Molecular Property Prediction (MPP) is vital for drug discovery, crop protection, and environmental science. Over the last decades, diverse computational techniques have been developed, from using simple physical and chemical properties and molecular fingerprints in statistical models and classical machine learning to advanced deep learning approaches. In this review, we aim to distill insights from current research on employing transformer models for MPP. We analyze the currently available models and explore key questions that arise when training and fine-tuning a transformer model for MPP. These questions encompass the choice and scale of the pre-training data, optimal architecture selections, and promising pre-training objectives. Our analysis highlights areas not yet covered in current research, inviting further exploration to enhance the field's understanding. Additionally, we address the challenges in comparing different models, emphasizing the need for standardized data splitting and robust statistical analysis.
4/8/2024
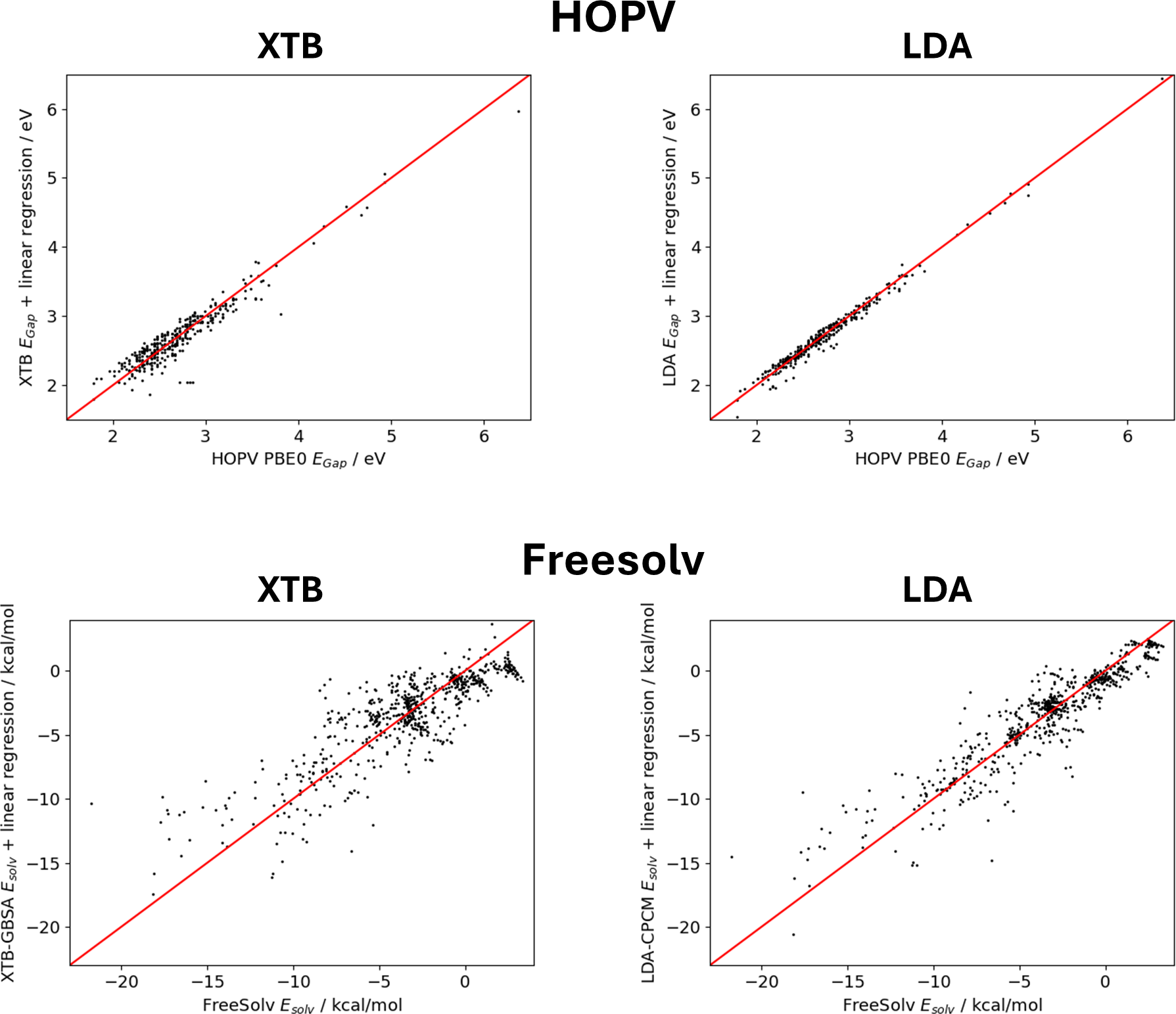
Transfer Learning for Molecular Property Predictions from Small Data Sets
Thorren Kirschbaum, Annika Bande
0
0
Machine learning has emerged as a new tool in chemistry to bypass expensive experiments or quantum-chemical calculations, for example, in high-throughput screening applications. However, many machine learning studies rely on small data sets, making it difficult to efficiently implement powerful deep learning architectures such as message passing neural networks. In this study, we benchmark common machine learning models for the prediction of molecular properties on small data sets, for which the best results are obtained with the message passing neural network PaiNN, as well as SOAP molecular descriptors concatenated to a set of simple molecular descriptors tailored to gradient boosting with regression trees. To further improve the predictive capabilities of PaiNN, we present a transfer learning strategy that uses large data sets to pre-train the respective models and allows to obtain more accurate models after fine-tuning on the original data sets. The pre-training labels are obtained from computationally cheap ab initio or semi-empirical models and corrected by simple linear regression on the target data set to obtain labels that are close to those of the original data. This strategy is tested on the Harvard Oxford Photovoltaics data set (HOPV, HOMO-LUMO-gaps), for which excellent results are obtained, and on the Freesolv data set (solvation energies), where this method is unsuccessful due to a complex underlying learning task and the dissimilar methods used to obtain pre-training and fine-tuning labels. Finally, we find that the final training results do not improve monotonically with the size of the pre-training data set, but pre-training with fewer data points can lead to more biased pre-trained models and higher accuracy after fine-tuning.
4/23/2024