The Role of Model Architecture and Scale in Predicting Molecular Properties: Insights from Fine-Tuning RoBERTa, BART, and LLaMA
2405.00949
0
0
📈
Abstract
This study introduces a systematic framework to compare the efficacy of Large Language Models (LLMs) for fine-tuning across various cheminformatics tasks. Employing a uniform training methodology, we assessed three well-known models-RoBERTa, BART, and LLaMA-on their ability to predict molecular properties using the Simplified Molecular Input Line Entry System (SMILES) as a universal molecular representation format. Our comparative analysis involved pre-training 18 configurations of these models, with varying parameter sizes and dataset scales, followed by fine-tuning them on six benchmarking tasks from DeepChem. We maintained consistent training environments across models to ensure reliable comparisons. This approach allowed us to assess the influence of model type, size, and training dataset size on model performance. Specifically, we found that LLaMA-based models generally offered the lowest validation loss, suggesting their superior adaptability across tasks and scales. However, we observed that absolute validation loss is not a definitive indicator of model performance - contradicts previous research - at least for fine-tuning tasks: instead, model size plays a crucial role. Through rigorous replication and validation, involving multiple training and fine-tuning cycles, our study not only delineates the strengths and limitations of each model type but also provides a robust methodology for selecting the most suitable LLM for specific cheminformatics applications. This research underscores the importance of considering model architecture and dataset characteristics in deploying AI for molecular property prediction, paving the way for more informed and effective utilization of AI in drug discovery and related fields.
Create account to get full access
Overview
- This study compares the effectiveness of different large language models (LLMs) for fine-tuning on cheminformatics tasks.
- The researchers trained three well-known LLMs (RoBERTa, BART, and LLaMA) using a consistent methodology and evaluated their performance on six benchmark tasks from DeepChem.
- The goal was to assess the influence of model type, size, and training dataset size on model performance for molecular property prediction.
Plain English Explanation
The researchers in this study wanted to understand how effective different large language models (LLMs) are at learning to predict properties of molecules. They took three popular LLMs - RoBERTa, BART, and LLaMA - and trained them in a consistent way, then tested them on a set of standard cheminformatics tasks.
Cheminformatics is the field of using computers to work with chemical information, like predicting the properties of new drug candidates. The researchers used a common way of representing molecules called SMILES as the input to the LLMs. They trained 18 different configurations of the models, varying the model size and the amount of training data, to see how those factors affected the models' performance.
The key finding was that the LLaMA-based models generally had the lowest validation loss, suggesting they were the most adaptable across the different tasks and data scales. However, the researchers also found that the absolute validation loss wasn't the only important factor - the size of the model also played a crucial role in its performance.
This research provides a solid methodology for selecting the best LLM for cheminformatics applications, taking into account both the model architecture and the characteristics of the dataset. It highlights the importance of considering these factors when using AI for tasks like drug discovery.
Technical Explanation
The researchers in this study introduced a systematic framework to compare the performance of different large language models (LLMs) on a variety of cheminformatics tasks. They focused on three well-known LLM architectures: RoBERTa, BART, and LLaMA.
The researchers employed a uniform training methodology to assess these models' ability to predict molecular properties using the Simplified Molecular Input Line Entry System (SMILES) as a universal molecular representation format. They pre-trained 18 different configurations of the models, varying the parameter size and dataset scale, and then fine-tuned them on six benchmark tasks from the DeepChem library.
By maintaining consistent training environments across the models, the researchers were able to reliably compare their performance. This allowed them to evaluate the influence of model type, size, and training dataset size on the models' predictive capabilities.
The key finding was that the LLaMA-based models generally offered the lowest validation loss, suggesting their superior adaptability across tasks and scales. However, the researchers also observed that absolute validation loss is not a definitive indicator of model performance for fine-tuning tasks. Instead, they found that model size plays a crucial role in determining the models' effectiveness.
Through rigorous replication and validation, involving multiple training and fine-tuning cycles, the study not only delineates the strengths and limitations of each model type but also provides a robust methodology for selecting the most suitable LLM for specific cheminformatics applications.
Critical Analysis
The researchers in this study have provided a comprehensive and systematic framework for evaluating the performance of different LLMs on cheminformatics tasks. The use of a consistent training methodology and a diverse set of benchmark tasks helps to ensure the reliability and generalizability of their findings.
However, the study does not address some potential limitations or areas for further research. For example, the researchers only focused on three LLM architectures, and it would be interesting to see how other models, such as MolTailor, perform in this context.
Additionally, the study does not delve into the specific reasons why the LLaMA-based models exhibited superior adaptability. It would be valuable to understand the underlying architectural or training-related factors that contributed to this performance advantage.
Furthermore, the researchers acknowledge that absolute validation loss is not the only relevant metric for evaluating model performance. It would be helpful to explore other evaluation metrics, such as task-specific scores or real-world application-oriented measures, to gain a more holistic understanding of the models' capabilities.
Despite these potential areas for further exploration, the researchers have made a significant contribution to the field of cheminformatics by providing a robust methodology for selecting appropriate LLMs for various tasks. This research underscores the importance of considering both model architecture and dataset characteristics when deploying AI for molecular property prediction and related applications.
Conclusion
This study introduces a systematic framework for comparing the effectiveness of different large language models (LLMs) in fine-tuning on cheminformatics tasks. The researchers trained three well-known LLMs - RoBERTa, BART, and LLaMA - using a consistent methodology and evaluated their performance on six benchmark tasks from the DeepChem library.
The key finding was that the LLaMA-based models generally exhibited the lowest validation loss, suggesting their superior adaptability across tasks and scales. However, the researchers also discovered that absolute validation loss is not the sole determinant of model performance; model size also plays a crucial role.
By providing a robust and replicable methodology for assessing LLM performance, this research paves the way for more informed and effective utilization of AI in drug discovery and other cheminformatics applications. It underscores the importance of considering both model architecture and dataset characteristics when deploying AI for molecular property prediction, helping researchers and practitioners make more informed decisions about the most suitable LLMs for their specific needs.
This summary was produced with help from an AI and may contain inaccuracies - check out the links to read the original source documents!
Related Papers

Ensemble Model With Bert,Roberta and Xlnet For Molecular property prediction
Junling Hu
0
0
This paper presents a novel approach for predicting molecular properties with high accuracy without the need for extensive pre-training. Employing ensemble learning and supervised fine-tuning of BERT, RoBERTa, and XLNet, our method demonstrates significant effectiveness compared to existing advanced models. Crucially, it addresses the issue of limited computational resources faced by experimental groups, enabling them to accurately predict molecular properties. This innovation provides a cost-effective and resource-efficient solution, potentially advancing further research in the molecular domain.
6/12/2024

Can Large Language Models Understand Molecules?
Shaghayegh Sadeghi, Alan Bui, Ali Forooghi, Jianguo Lu, Alioune Ngom
0
0
Purpose: Large Language Models (LLMs) like GPT (Generative Pre-trained Transformer) from OpenAI and LLaMA (Large Language Model Meta AI) from Meta AI are increasingly recognized for their potential in the field of cheminformatics, particularly in understanding Simplified Molecular Input Line Entry System (SMILES), a standard method for representing chemical structures. These LLMs also have the ability to decode SMILES strings into vector representations. Method: We investigate the performance of GPT and LLaMA compared to pre-trained models on SMILES in embedding SMILES strings on downstream tasks, focusing on two key applications: molecular property prediction and drug-drug interaction prediction. Results: We find that SMILES embeddings generated using LLaMA outperform those from GPT in both molecular property and DDI prediction tasks. Notably, LLaMA-based SMILES embeddings show results comparable to pre-trained models on SMILES in molecular prediction tasks and outperform the pre-trained models for the DDI prediction tasks. Conclusion: The performance of LLMs in generating SMILES embeddings shows great potential for further investigation of these models for molecular embedding. We hope our study bridges the gap between LLMs and molecular embedding, motivating additional research into the potential of LLMs in the molecular representation field. GitHub: https://github.com/sshaghayeghs/LLaMA-VS-GPT
5/22/2024
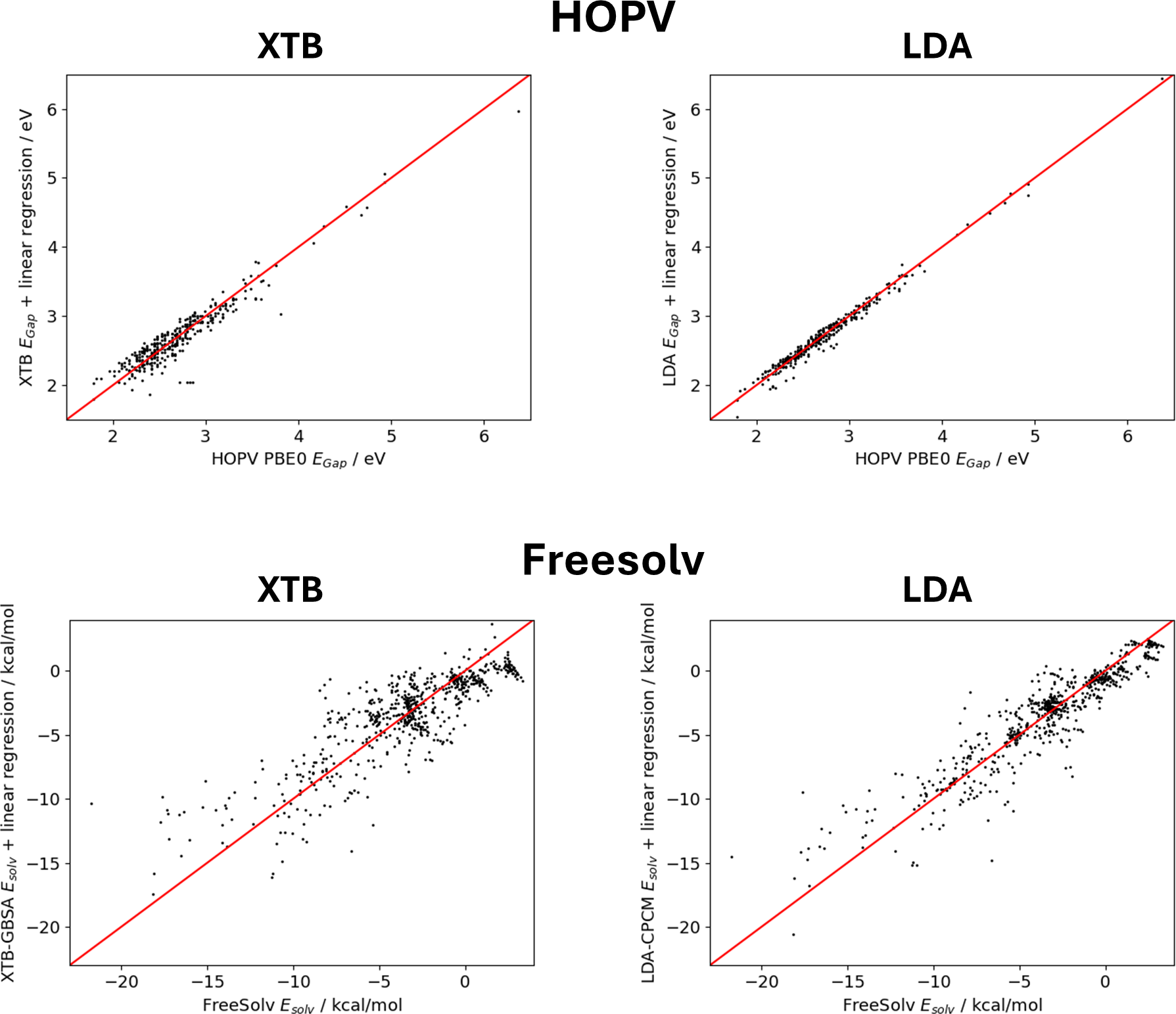
Transfer Learning for Molecular Property Predictions from Small Data Sets
Thorren Kirschbaum, Annika Bande
0
0
Machine learning has emerged as a new tool in chemistry to bypass expensive experiments or quantum-chemical calculations, for example, in high-throughput screening applications. However, many machine learning studies rely on small data sets, making it difficult to efficiently implement powerful deep learning architectures such as message passing neural networks. In this study, we benchmark common machine learning models for the prediction of molecular properties on small data sets, for which the best results are obtained with the message passing neural network PaiNN, as well as SOAP molecular descriptors concatenated to a set of simple molecular descriptors tailored to gradient boosting with regression trees. To further improve the predictive capabilities of PaiNN, we present a transfer learning strategy that uses large data sets to pre-train the respective models and allows to obtain more accurate models after fine-tuning on the original data sets. The pre-training labels are obtained from computationally cheap ab initio or semi-empirical models and corrected by simple linear regression on the target data set to obtain labels that are close to those of the original data. This strategy is tested on the Harvard Oxford Photovoltaics data set (HOPV, HOMO-LUMO-gaps), for which excellent results are obtained, and on the Freesolv data set (solvation energies), where this method is unsuccessful due to a complex underlying learning task and the dissimilar methods used to obtain pre-training and fine-tuning labels. Finally, we find that the final training results do not improve monotonically with the size of the pre-training data set, but pre-training with fewer data points can lead to more biased pre-trained models and higher accuracy after fine-tuning.
4/23/2024
Explainable Molecular Property Prediction: Aligning Chemical Concepts with Predictions via Language Models
Zhenzhong Wang, Zehui Lin, Wanyu Lin, Ming Yang, Minggang Zeng, Kay Chen Tan
0
0
Providing explainable molecule property predictions is critical for many scientific domains, such as drug discovery and material science. Though transformer-based language models have shown great potential in accurate molecular property prediction, they neither provide chemically meaningful explanations nor faithfully reveal the molecular structure-property relationships. In this work, we develop a new framework for explainable molecular property prediction based on language models, dubbed as Lamole, which can provide chemical concepts-aligned explanations. We first leverage a designated molecular representation -- the Group SELFIES -- as it can provide chemically meaningful semantics. Because attention mechanisms in Transformers can inherently capture relationships within the input, we further incorporate the attention weights and gradients together to generate explanations for capturing the functional group interactions. We then carefully craft a marginal loss to explicitly optimize the explanations to be able to align with the chemists' annotations. We bridge the manifold hypothesis with the elaborated marginal loss to prove that the loss can align the explanations with the tangent space of the data manifold, leading to concept-aligned explanations. Experimental results over six mutagenicity datasets and one hepatotoxicity dataset demonstrate Lamole can achieve comparable classification accuracy and boost the explanation accuracy by up to 14.8%, being the state-of-the-art in explainable molecular property prediction.
6/4/2024