Transfer Learning for Molecular Property Predictions from Small Data Sets
2404.13393
0
0
Abstract
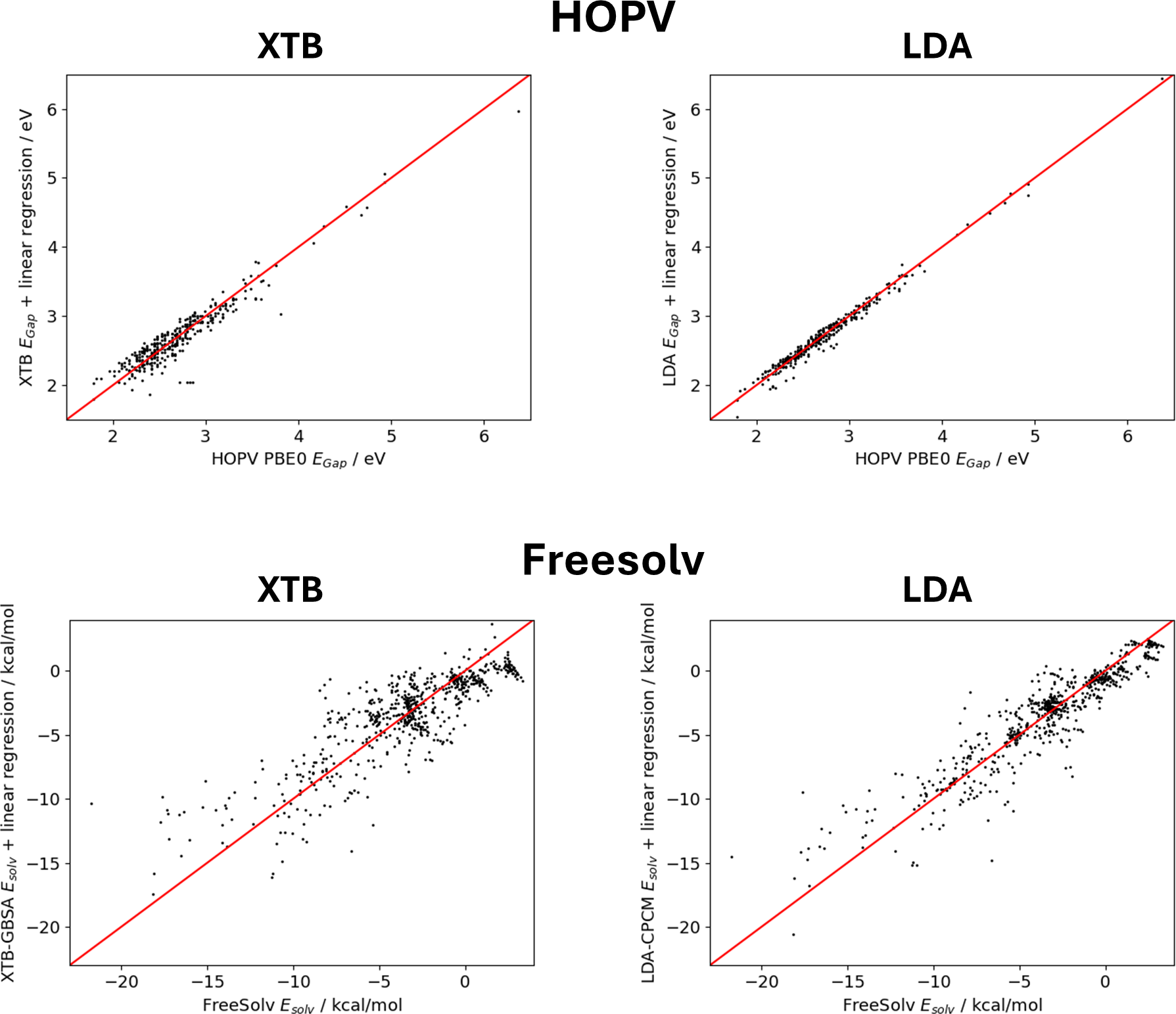
Machine learning has emerged as a new tool in chemistry to bypass expensive experiments or quantum-chemical calculations, for example, in high-throughput screening applications. However, many machine learning studies rely on small data sets, making it difficult to efficiently implement powerful deep learning architectures such as message passing neural networks. In this study, we benchmark common machine learning models for the prediction of molecular properties on small data sets, for which the best results are obtained with the message passing neural network PaiNN, as well as SOAP molecular descriptors concatenated to a set of simple molecular descriptors tailored to gradient boosting with regression trees. To further improve the predictive capabilities of PaiNN, we present a transfer learning strategy that uses large data sets to pre-train the respective models and allows to obtain more accurate models after fine-tuning on the original data sets. The pre-training labels are obtained from computationally cheap ab initio or semi-empirical models and corrected by simple linear regression on the target data set to obtain labels that are close to those of the original data. This strategy is tested on the Harvard Oxford Photovoltaics data set (HOPV, HOMO-LUMO-gaps), for which excellent results are obtained, and on the Freesolv data set (solvation energies), where this method is unsuccessful due to a complex underlying learning task and the dissimilar methods used to obtain pre-training and fine-tuning labels. Finally, we find that the final training results do not improve monotonically with the size of the pre-training data set, but pre-training with fewer data points can lead to more biased pre-trained models and higher accuracy after fine-tuning.
Create account to get full access
Overview
- This paper investigates using transfer learning to improve the accuracy of machine learning models for predicting molecular properties from small datasets.
- The researchers explore leveraging pre-trained models and fine-tuning them on target tasks to overcome the limited data challenge in molecular property prediction.
- The paper compares the performance of transfer learning approaches to models trained from scratch on small datasets.
Plain English Explanation
Predicting the properties of molecules is an important task in fields like chemistry and drug discovery. However, building accurate machine learning models for this can be challenging because the available datasets are often small. Transfer Learning for Molecular Property Predictions from Small Data Sets explores a potential solution to this problem - using transfer learning.
Transfer learning involves taking a model that has been trained on a large, general dataset and fine-tuning it on the specific task you care about, which in this case is predicting molecular properties. The idea is that the model will have already learned useful features and patterns from the large dataset that can be leveraged for the target task, even if the data is limited.
The researchers in this paper compare the performance of models trained from scratch on small datasets to those that use transfer learning. They find that the transfer learning approach can significantly improve accuracy, especially when the target dataset is very small. This suggests that transfer learning could be a powerful technique for tackling the data scarcity challenge in molecular property prediction.
Technical Explanation
The paper investigates using transfer learning to improve the accuracy of machine learning models for predicting molecular properties from small datasets. The researchers explore leveraging pre-trained models, such as those trained on large general chemical datasets, and fine-tuning them on target property prediction tasks.
The experimental setup compares the performance of transfer learning approaches to models trained from scratch on small datasets. The transfer learning models start with weights from a pre-trained model and then fine-tune these weights on the target task. The baseline models are trained solely on the limited target dataset.
The results show that transfer learning can significantly boost the accuracy of molecular property prediction, especially when the target dataset is very small. The transfer learning models outperformed the baseline models trained from scratch by a large margin in these data-scarce regimes.
The paper provides insights into how the choice of pre-trained model, fine-tuning hyperparameters, and target task characteristics impact the effectiveness of transfer learning. It also discusses potential limitations, such as the transferability of learned representations across different molecular property prediction tasks.
Critical Analysis
The paper provides a thorough investigation of using transfer learning to address the data scarcity challenge in molecular property prediction. The results demonstrate the power of this approach, especially for small target datasets. However, the authors acknowledge some caveats and limitations.
One limitation is that the transferability of learned representations may vary across different molecular property prediction tasks. The effectiveness of transfer learning could depend on the relatedness between the pre-training and target tasks. Further research is needed to understand the optimal selection of pre-trained models for specific target tasks.
Additionally, the paper does not explore the generalization of the transfer learning approach to truly novel molecules outside the training distribution. While the results are promising for interpolation within known chemical space, the ability to extrapolate to new molecular structures remains an open question.
Advancing Extrapolative Predictions of Material Properties Through Learning and Lightweight Geometric Deep Learning for Molecular Modelling and Catalyst Design highlight the challenges of extrapolation in molecular property prediction that could be further investigated in the context of transfer learning.
Overall, this paper makes a valuable contribution by demonstrating the effectiveness of transfer learning for improving molecular property prediction from small datasets. The insights and limitations provide a solid foundation for future research in this area.
Conclusion
This paper showcases the potential of transfer learning to address the data scarcity challenge in molecular property prediction. By leveraging pre-trained models and fine-tuning them on target tasks, the researchers were able to significantly improve the accuracy of property prediction, especially when working with small datasets.
The results suggest that transfer learning could be a powerful technique for advancing the field of computational chemistry and accelerating the discovery of new materials and drugs. By reducing the data requirements for accurate property prediction, transfer learning can enable more efficient and cost-effective research and development.
Learning Quantum Properties from Short-Range Correlations and Multimodal Learning for Predicting Molecular Properties: A Framework-Based Approach highlight complementary approaches that could be combined with transfer learning to further enhance molecular property prediction capabilities.
Overall, this paper provides valuable insights and a promising direction for future research in the application of machine learning to problems in chemistry and materials science.
This summary was produced with help from an AI and may contain inaccuracies - check out the links to read the original source documents!
Related Papers

Transformers for molecular property prediction: Lessons learned from the past five years
Afnan Sultan, Jochen Sieg, Miriam Mathea, Andrea Volkamer
0
0
Molecular Property Prediction (MPP) is vital for drug discovery, crop protection, and environmental science. Over the last decades, diverse computational techniques have been developed, from using simple physical and chemical properties and molecular fingerprints in statistical models and classical machine learning to advanced deep learning approaches. In this review, we aim to distill insights from current research on employing transformer models for MPP. We analyze the currently available models and explore key questions that arise when training and fine-tuning a transformer model for MPP. These questions encompass the choice and scale of the pre-training data, optimal architecture selections, and promising pre-training objectives. Our analysis highlights areas not yet covered in current research, inviting further exploration to enhance the field's understanding. Additionally, we address the challenges in comparing different models, emphasizing the need for standardized data splitting and robust statistical analysis.
4/8/2024
🔮
From Molecules to Materials: Pre-training Large Generalizable Models for Atomic Property Prediction
Nima Shoghi, Adeesh Kolluru, John R. Kitchin, Zachary W. Ulissi, C. Lawrence Zitnick, Brandon M. Wood
0
0
Foundation models have been transformational in machine learning fields such as natural language processing and computer vision. Similar success in atomic property prediction has been limited due to the challenges of training effective models across multiple chemical domains. To address this, we introduce Joint Multi-domain Pre-training (JMP), a supervised pre-training strategy that simultaneously trains on multiple datasets from different chemical domains, treating each dataset as a unique pre-training task within a multi-task framework. Our combined training dataset consists of $sim$120M systems from OC20, OC22, ANI-1x, and Transition-1x. We evaluate performance and generalization by fine-tuning over a diverse set of downstream tasks and datasets including: QM9, rMD17, MatBench, QMOF, SPICE, and MD22. JMP demonstrates an average improvement of 59% over training from scratch, and matches or sets state-of-the-art on 34 out of 40 tasks. Our work highlights the potential of pre-training strategies that utilize diverse data to advance property prediction across chemical domains, especially for low-data tasks. Please visit https://nima.sh/jmp for further information.
5/7/2024
📈
The Role of Model Architecture and Scale in Predicting Molecular Properties: Insights from Fine-Tuning RoBERTa, BART, and LLaMA
Lee Youngmin, Lang S. I. D. Andrew, Cai Duoduo, Wheat R. Stephen
0
0
This study introduces a systematic framework to compare the efficacy of Large Language Models (LLMs) for fine-tuning across various cheminformatics tasks. Employing a uniform training methodology, we assessed three well-known models-RoBERTa, BART, and LLaMA-on their ability to predict molecular properties using the Simplified Molecular Input Line Entry System (SMILES) as a universal molecular representation format. Our comparative analysis involved pre-training 18 configurations of these models, with varying parameter sizes and dataset scales, followed by fine-tuning them on six benchmarking tasks from DeepChem. We maintained consistent training environments across models to ensure reliable comparisons. This approach allowed us to assess the influence of model type, size, and training dataset size on model performance. Specifically, we found that LLaMA-based models generally offered the lowest validation loss, suggesting their superior adaptability across tasks and scales. However, we observed that absolute validation loss is not a definitive indicator of model performance - contradicts previous research - at least for fine-tuning tasks: instead, model size plays a crucial role. Through rigorous replication and validation, involving multiple training and fine-tuning cycles, our study not only delineates the strengths and limitations of each model type but also provides a robust methodology for selecting the most suitable LLM for specific cheminformatics applications. This research underscores the importance of considering model architecture and dataset characteristics in deploying AI for molecular property prediction, paving the way for more informed and effective utilization of AI in drug discovery and related fields.
5/3/2024
🌀
New!From molecules to scaffolds to functional groups: building context-dependent molecular representation via multi-channel learning
Yue Wan, Jialu Wu, Tingjun Hou, Chang-Yu Hsieh, Xiaowei Jia
0
0
Reliable molecular property prediction is essential for various scientific endeavors and industrial applications, such as drug discovery. However, the data scarcity, combined with the highly non-linear causal relationships between physicochemical and biological properties and conventional molecular featurization schemes, complicates the development of robust molecular machine learning models. Self-supervised learning (SSL) has emerged as a popular solution, utilizing large-scale, unannotated molecular data to learn a foundational representation of chemical space that might be advantageous for downstream tasks. Yet, existing molecular SSL methods largely overlook chemical knowledge, including molecular structure similarity, scaffold composition, and the context-dependent aspects of molecular properties when operating over the chemical space. They also struggle to learn the subtle variations in structure-activity relationship. This paper introduces a novel pre-training framework that learns robust and generalizable chemical knowledge. It leverages the structural hierarchy within the molecule, embeds them through distinct pre-training tasks across channels, and aggregates channel information in a task-specific manner during fine-tuning. Our approach demonstrates competitive performance across various molecular property benchmarks and offers strong advantages in particularly challenging yet ubiquitous scenarios like activity cliffs.
7/2/2024