Machine learning Hubbard parameters with equivariant neural networks

0
Sign in to get full access
Overview
- This paper explores the use of equivariant neural networks to model Hubbard parameters, which are important for understanding the electronic properties of materials.
- The key ideas include using occupation matrices as model inputs, designing equivariant descriptors to capture the symmetry properties of the system, and training the neural network to predict the Hubbard parameters.
- The results demonstrate the effectiveness of this approach compared to previous methods, with the equivariant neural network achieving improved accuracy in predicting the Hubbard parameters.
Plain English Explanation
In this paper, the researchers developed a new machine learning technique to better understand the electronic properties of materials. They focused on modeling a set of parameters called Hubbard parameters, which are important for describing the behavior of electrons in these materials.
The researchers used a type of neural network called an equivariant neural network, which is designed to capture the symmetry properties of the system. Instead of feeding the neural network raw data about the material, they used a special representation called an "occupation matrix" that encodes information about the electrons in the system.
By using this equivariant neural network and the occupation matrix representation, the researchers were able to achieve better accuracy in predicting the Hubbard parameters compared to previous methods. This is an important step forward in using machine learning to model the electronic structure of materials, which could have implications for designing new materials with desired electronic properties.
Technical Explanation
The key elements of this paper are:
-
Occupation matrices as the model inputs: The researchers used occupation matrices, which encode information about the occupation of electronic states in the material, as the input to their neural network. This representation captures important symmetry properties of the system.
-
Equivariant descriptors: To further exploit the symmetry properties of the system, the researchers designed a set of equivariant descriptors that transform in a specific way under the symmetry operations of the material. These descriptors were used as additional inputs to the neural network.
-
Equivariant neural network architecture: The neural network architecture was designed to be equivariant, meaning that it respects the symmetry properties of the input data. This was achieved through the use of equivariant layers in the network.
The results show that this equivariant neural network approach outperforms previous methods for predicting the Hubbard parameters, demonstrating the effectiveness of exploiting the symmetry properties of the system. This work builds on previous research in using machine learning for electronic structure and equivariant neural networks.
Critical Analysis
The paper provides a thorough evaluation of the proposed method, including comparisons to previous approaches and an analysis of the impact of different components of the model. However, there are a few potential limitations and areas for further research:
- The method is demonstrated on a limited set of materials, and it would be important to evaluate its performance on a wider range of systems to better understand its general applicability.
- The paper does not discuss how the method would scale to larger, more complex materials, which is an important consideration for practical applications.
- While the equivariant neural network architecture is a key innovation, the paper does not provide a detailed analysis of how the different equivariant layers contribute to the overall performance.
Overall, this work represents an important step forward in using machine learning to model the electronic structure of materials, and the equivariant neural network approach could have broader applications in materials science and chemistry.
Conclusion
This paper demonstrates the effectiveness of using equivariant neural networks to model Hubbard parameters, which are crucial for understanding the electronic properties of materials. By exploiting the symmetry properties of the system through the use of occupation matrices and equivariant descriptors, the researchers were able to achieve improved accuracy in predicting the Hubbard parameters compared to previous methods.
This work has important implications for the use of machine learning in materials science and could contribute to the development of new materials with tailored electronic properties. The equivariant neural network approach presented in this paper could also have broader applications in modeling the electronic structure of molecules and materials and active learning for effective Hamiltonians.
This summary was produced with help from an AI and may contain inaccuracies - check out the links to read the original source documents!
Related Papers
0
Machine learning Hubbard parameters with equivariant neural networks
Martin Uhrin, Austin Zadoks, Luca Binci, Nicola Marzari, Iurii Timrov
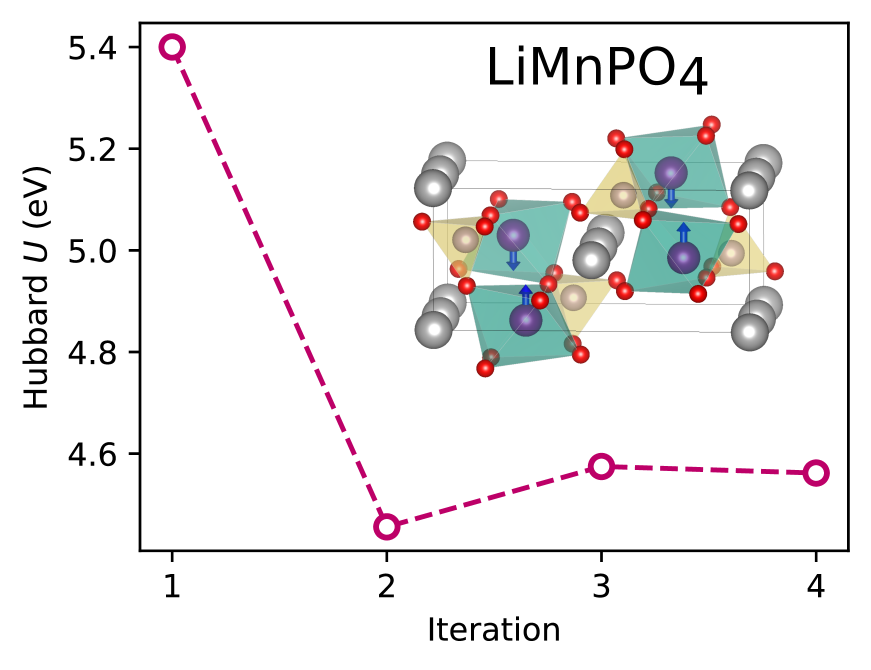
Density-functional theory with extended Hubbard functionals (DFT+$U$+$V$) provides a robust framework to accurately describe complex materials containing transition-metal or rare-earth elements. It does so by mitigating self-interaction errors inherent to semi-local functionals which are particularly pronounced in systems with partially-filled $d$ and $f$ electronic states. However, achieving accuracy in this approach hinges upon the accurate determination of the on-site $U$ and inter-site $V$ Hubbard parameters. In practice, these are obtained either by semi-empirical tuning, requiring prior knowledge, or, more correctly, by using predictive but expensive first-principles calculations. Here, we present a machine learning model based on equivariant neural networks which uses atomic occupation matrices as descriptors, directly capturing the electronic structure, local chemical environment, and oxidation states of the system at hand. We target here the prediction of Hubbard parameters computed self-consistently with iterative linear-response calculations, as implemented in density-functional perturbation theory (DFPT), and structural relaxations. Remarkably, when trained on data from 11 materials spanning various crystal structures and compositions, our model achieves mean absolute relative errors of 3% and 5% for Hubbard $U$ and $V$ parameters, respectively. By circumventing computationally expensive DFT or DFPT self-consistent protocols, our model significantly expedites the prediction of Hubbard parameters with negligible computational overhead, while approaching the accuracy of DFPT. Moreover, owing to its robust transferability, the model facilitates accelerated materials discovery and design via high-throughput calculations, with relevance for various technological applications.
Read more6/5/2024
💬
0
Universal Machine Learning Kohn-Sham Hamiltonian for Materials
Yang Zhong, Hongyu Yu, Jihui Yang, Xingyu Guo, Hongjun Xiang, Xingao Gong
While density functional theory (DFT) serves as a prevalent computational approach in electronic structure calculations, its computational demands and scalability limitations persist. Recently, leveraging neural networks to parameterize the Kohn-Sham DFT Hamiltonian has emerged as a promising avenue for accelerating electronic structure computations. Despite advancements, challenges such as the necessity for computing extensive DFT training data to explore each new system and the complexity of establishing accurate ML models for multi-elemental materials still exist. Addressing these hurdles, this study introduces a universal electronic Hamiltonian model trained on Hamiltonian matrices obtained from first-principles DFT calculations of nearly all crystal structures on the Materials Project. We demonstrate its generality in predicting electronic structures across the whole periodic table, including complex multi-elemental systems, solid-state electrolytes, Moir'e twisted bilayer heterostructure, and metal-organic frameworks (MOFs). Moreover, we utilize the universal model to conduct high-throughput calculations of electronic structures for crystals in GeNOME datasets, identifying 3,940 crystals with direct band gaps and 5,109 crystals with flat bands. By offering a reliable efficient framework for computing electronic properties, this universal Hamiltonian model lays the groundwork for advancements in diverse fields, such as easily providing a huge data set of electronic structures and also making the materials design across the whole periodic table possible.
Read more4/16/2024
0
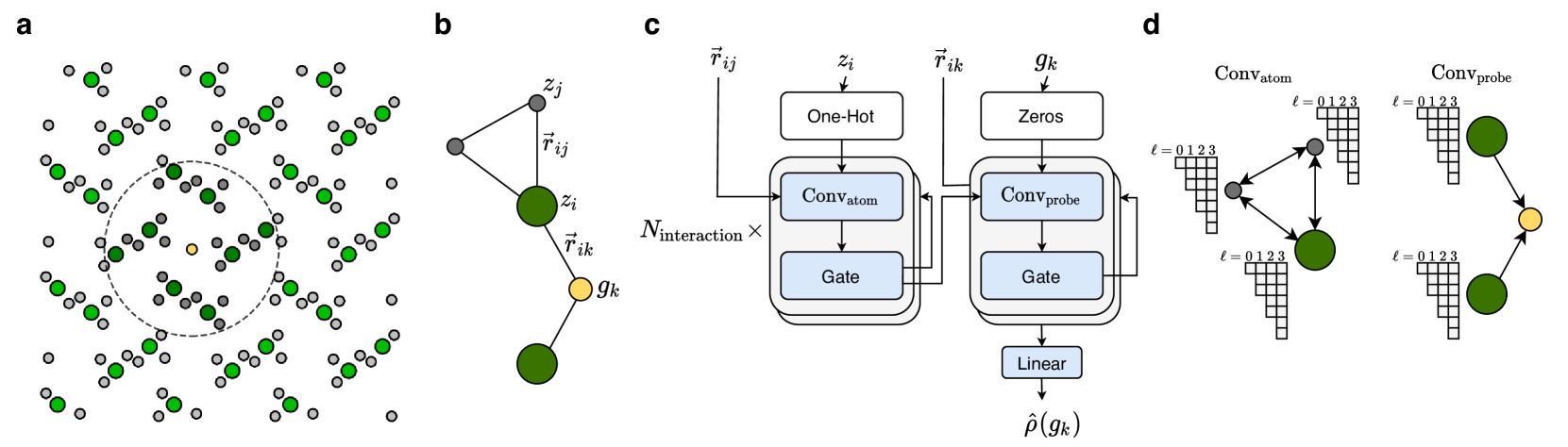
Higher-Order Equivariant Neural Networks for Charge Density Prediction in Materials
Teddy Koker, Keegan Quigley, Eric Taw, Kevin Tibbetts, Lin Li
The calculation of electron density distribution using density functional theory (DFT) in materials and molecules is central to the study of their quantum and macro-scale properties, yet accurate and efficient calculation remains a long-standing challenge. We introduce ChargE3Net, an E(3)-equivariant graph neural network for predicting electron density in atomic systems. ChargE3Net enables the learning of higher-order equivariant feature to achieve high predictive accuracy and model expressivity. We show that ChargE3Net exceeds the performance of prior work on diverse sets of molecules and materials. When trained on the massive dataset of over 100K materials in the Materials Project database, our model is able to capture the complexity and variability in the data, leading to a significant 26.7% reduction in self-consistent iterations when used to initialize DFT calculations on unseen materials. Furthermore, we show that non-self-consistent DFT calculations using our predicted charge densities yield near-DFT performance on electronic and thermodynamic property prediction at a fraction of the computational cost. Further analysis attributes the greater predictive accuracy to improved modeling of systems with high angular variations. These results illuminate a pathway towards a machine learning-accelerated ab initio calculations for materials discovery.
Read more5/15/2024
0
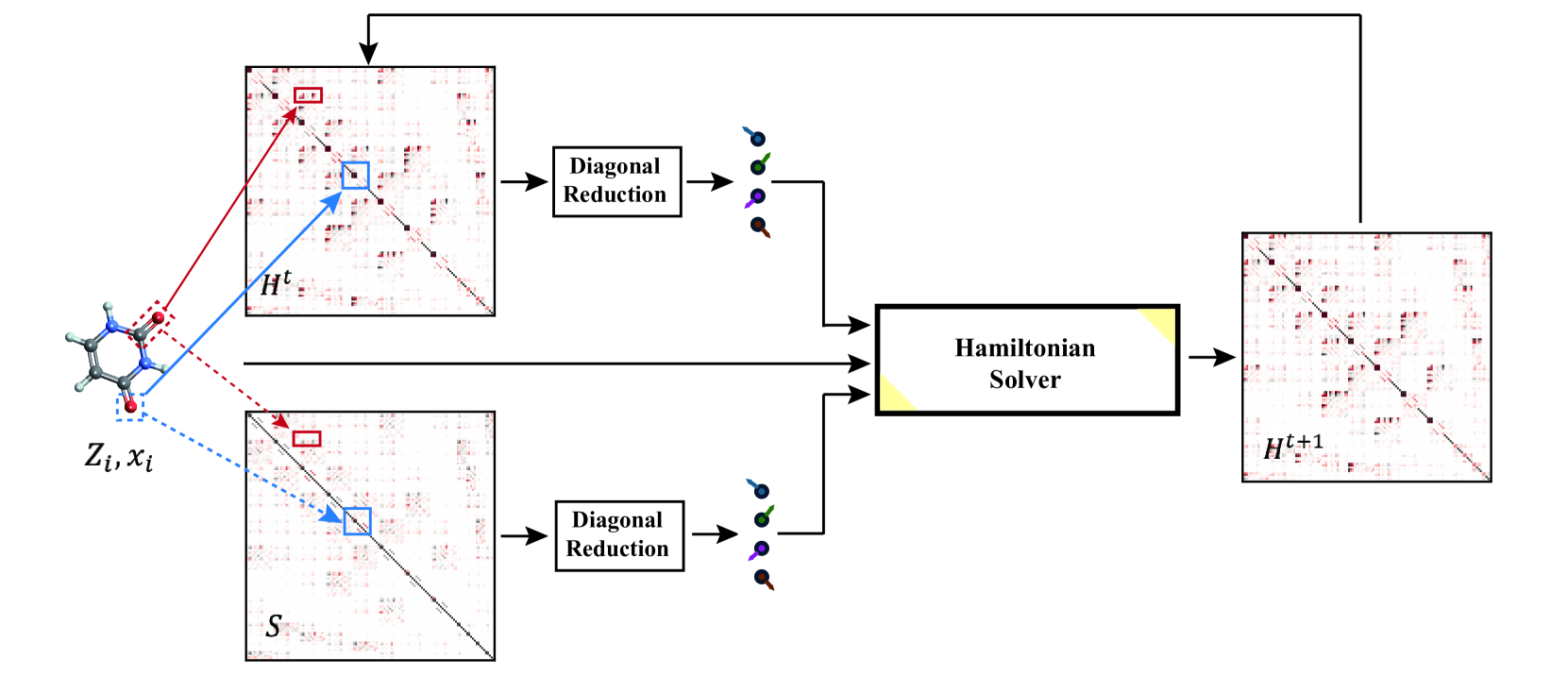
Infusing Self-Consistency into Density Functional Theory Hamiltonian Prediction via Deep Equilibrium Models
Zun Wang, Chang Liu, Nianlong Zou, He Zhang, Xinran Wei, Lin Huang, Lijun Wu, Bin Shao
In this study, we introduce a unified neural network architecture, the Deep Equilibrium Density Functional Theory Hamiltonian (DEQH) model, which incorporates Deep Equilibrium Models (DEQs) for predicting Density Functional Theory (DFT) Hamiltonians. The DEQH model inherently captures the self-consistency nature of Hamiltonian, a critical aspect often overlooked by traditional machine learning approaches for Hamiltonian prediction. By employing DEQ within our model architecture, we circumvent the need for DFT calculations during the training phase to introduce the Hamiltonian's self-consistency, thus addressing computational bottlenecks associated with large or complex systems. We propose a versatile framework that combines DEQ with off-the-shelf machine learning models for predicting Hamiltonians. When benchmarked on the MD17 and QH9 datasets, DEQHNet, an instantiation of the DEQH framework, has demonstrated a significant improvement in prediction accuracy. Beyond a predictor, the DEQH model is a Hamiltonian solver, in the sense that it uses the fixed-point solving capability of the deep equilibrium model to iteratively solve for the Hamiltonian. Ablation studies of DEQHNet further elucidate the network's effectiveness, offering insights into the potential of DEQ-integrated networks for Hamiltonian learning.
Read more6/7/2024