Material Property Prediction using Graphs based on Generically Complete Isometry Invariants

0
🔮
Sign in to get full access
Overview
- The paper discusses a new approach to predicting material properties using a simpler graph representation called the Distribution Graph.
- The key innovation is the use of the Pointwise Distance Distribution (PDD) to distinguish between different crystal structures, which resolves the ambiguity in conventional crystal representations.
- The Distribution Graph based on PDD outperforms state-of-the-art graph neural network models like CGCNN and ALIGNN on materials property prediction tasks.
Plain English Explanation
The structure-property hypothesis states that the properties of all materials are determined by their underlying crystal structure. However, traditional ways of representing crystal structures were often ambiguous, leading to errors in property predictions.
The authors introduce the Pointwise Distance Distribution (PDD), which can unambiguously distinguish between different crystal structures, even in the world's largest database of real materials. This breakthrough allowed them to create a simpler graph representation called the Distribution Graph, with fewer vertices than previous graph-based models.
When applied to materials property prediction tasks, the Distribution Graph achieved better performance than state-of-the-art graph neural networks like CGCNN and ALIGNN, while using a more compact graph structure. The authors also provide theoretical and experimental justification for their approach to selecting hyperparameters for the graph.
Technical Explanation
The core innovation in this work is the use of the Pointwise Distance Distribution (PDD) to create a graph representation of crystal structures called the Distribution Graph. The PDD can uniquely identify all periodic structures in large materials databases like the Cambridge Structural Database, resolving the ambiguity in conventional crystal representations.
The authors adapt the PDD to construct a simpler graph whose vertex set is no larger than the asymmetric unit of the crystal structure, in contrast to the full crystal graph used by models like CGCNN and ALIGNN.
When applied to materials property prediction tasks on the Materials Project and Jarvis-DFT datasets, the Distribution Graph achieved a 0.6%-12% reduction in mean absolute error compared to the crystal graph, while using 44%-88% fewer vertices. The authors provide theoretical justification for their hyperparameter selection methods, which are then validated experimentally.
Critical Analysis
The authors acknowledge that the PDD-based Distribution Graph may not be the optimal graph representation for all materials property prediction tasks, as the performance improvement over the crystal graph varies across different datasets and target properties.
Additionally, the authors do not provide a comprehensive comparison to other graph representation methods, such as AlphaFold's distance matrix-based approach. Further research could explore the relative strengths and weaknesses of these different graph representations.
It would also be valuable to understand the computational efficiency of the Distribution Graph compared to the crystal graph, as the reduced number of vertices may lead to faster training and inference times, which could be important for real-world applications.
Conclusion
This work introduces a novel graph representation for crystal structures called the Distribution Graph, which leverages the Pointwise Distance Distribution to unambiguously distinguish between different periodic structures. The simpler graph architecture outperforms state-of-the-art models on materials property prediction tasks, demonstrating the value of this new approach.
The authors' theoretical and experimental justification of their methods provides a strong foundation for further research into improving the efficiency and effectiveness of materials property prediction using graph-based models.
This summary was produced with help from an AI and may contain inaccuracies - check out the links to read the original source documents!
Related Papers
🔮
0
Material Property Prediction using Graphs based on Generically Complete Isometry Invariants
Jonathan Balasingham, Viktor Zamaraev, Vitaliy Kurlin
The structure-property hypothesis says that the properties of all materials are determined by an underlying crystal structure. The main obstacle was the ambiguity of conventional crystal representations based on incomplete or discontinuous descriptors that allow false negatives or false positives. This ambiguity was resolved by the ultra-fast Pointwise Distance Distribution (PDD), which distinguished all periodic structures in the world's largest collection of real materials (Cambridge Structural Database). The state-of-the-art results in property predictions were previously achieved by graph neural networks based on various graph representations of periodic crystals, including the Crystal Graph with vertices at all atoms in a crystal unit cell. This work adapts the Pointwise Distance Distribution for a simpler graph whose vertex set is not larger than the asymmetric unit of a crystal structure. The new Distribution Graph reduces mean-absolute-error by 0.6%-12% while having 44%-88% of the number of vertices when compared to the crystal graph when applied on the Materials Project and Jarvis-DFT datasets using CGCNN and ALIGNN. Methods for hyper-parameters selection for the graph are backed by the theoretical results of the Pointwise Distance Distribution and are then experimentally justified.
Read more5/8/2024

0
Accelerating Material Property Prediction using Generically Complete Isometry Invariants
Jonathan Balasingham, Viktor Zamaraev, Vitaliy Kurlin
Periodic material or crystal property prediction using machine learning has grown popular in recent years as it provides a computationally efficient replacement for classical simulation methods. A crucial first step for any of these algorithms is the representation used for a periodic crystal. While similar objects like molecules and proteins have a finite number of atoms and their representation can be built based upon a finite point cloud interpretation, periodic crystals are unbounded in size, making their representation more challenging. In the present work, we adapt the Pointwise Distance Distribution (PDD), a continuous and generically complete isometry invariant for periodic point sets, as a representation for our learning algorithm. The PDD distinguished all (more than 660 thousand) periodic crystals in the Cambridge Structural Database as purely periodic sets of points without atomic types. We develop a transformer model with a modified self-attention mechanism that combines PDD with compositional information via a spatial encoding method. This model is tested on the crystals of the Materials Project and Jarvis-DFT databases and shown to produce accuracy on par with state-of-the-art methods while being several times faster in both training and prediction time.
Read more5/8/2024
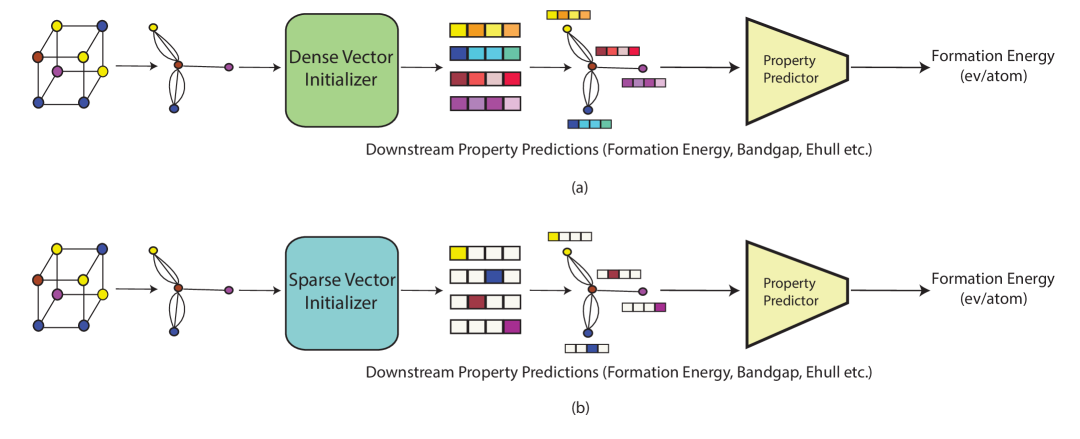
0
CrysAtom: Distributed Representation of Atoms for Crystal Property Prediction
Shrimon Mukherjee, Madhusudan Ghosh, Partha Basuchowdhuri
Application of artificial intelligence (AI) has been ubiquitous in the growth of research in the areas of basic sciences. Frequent use of machine learning (ML) and deep learning (DL) based methodologies by researchers has resulted in significant advancements in the last decade. These techniques led to notable performance enhancements in different tasks such as protein structure prediction, drug-target binding affinity prediction, and molecular property prediction. In material science literature, it is well-known that crystalline materials exhibit topological structures. Such topological structures may be represented as graphs and utilization of graph neural network (GNN) based approaches could help encoding them into an augmented representation space. Primarily, such frameworks adopt supervised learning techniques targeted towards downstream property prediction tasks on the basis of electronic properties (formation energy, bandgap, total energy, etc.) and crystalline structures. Generally, such type of frameworks rely highly on the handcrafted atom feature representations along with the structural representations. In this paper, we propose an unsupervised framework namely, CrysAtom, using untagged crystal data to generate dense vector representation of atoms, which can be utilized in existing GNN-based property predictor models to accurately predict important properties of crystals. Empirical results show that our dense representation embeds chemical properties of atoms and enhance the performance of the baseline property predictor models significantly.
Read more9/10/2024

0
Using GNN property predictors as molecule generators
F'elix Therrien, Edward H. Sargent, Oleksandr Voznyy
Graph neural networks (GNNs) have emerged as powerful tools to accurately predict materials and molecular properties in computational discovery pipelines. In this article, we exploit the invertible nature of these neural networks to directly generate molecular structures with desired electronic properties. Starting from a random graph or an existing molecule, we perform a gradient ascent while holding the GNN weights fixed in order to optimize its input, the molecular graph, towards the target property. Valence rules are enforced strictly through a judicious graph construction. The method relies entirely on the property predictor; no additional training is required on molecular structures. We demonstrate the application of this method by generating molecules with specific DFT-verified energy gaps and octanol-water partition coefficients (logP). Our approach hits target properties with rates comparable to or better than state-of-the-art generative models while consistently generating more diverse molecules.
Read more6/6/2024