Explainable Molecular Property Prediction: Aligning Chemical Concepts with Predictions via Language Models

0
Sign in to get full access
Overview
- This paper explores the challenge of providing explainable molecular property predictions, which is critical for the safe and responsible development of new chemicals and materials.
- The researchers propose an approach that aligns chemical concepts with model predictions using language models, aiming to make the predictions more interpretable and trustworthy.
- The method involves training a language model on chemical knowledge and then using it to extract relevant chemical concepts that explain the model's predictions for specific molecules.
Plain English Explanation
Predicting the properties of molecules is important for developing new chemicals and materials, but it can be difficult to understand how the prediction models work. This paper explores a way to make those predictions more interpretable.
The researchers used a language model - a type of AI that can understand and generate human language - to connect the predictions to chemical concepts that humans can understand. This allows them to explain why the model made a certain prediction for a given molecule.
For example, if the model predicts that a molecule will be a good electrical conductor, the language model could point to specific chemical properties, like the molecule's atomic structure or electron distribution, that support that prediction. This helps make the model's reasoning more transparent and trustworthy.
Technical Explanation
The key idea in this paper is to align chemical concepts with model predictions using language models. The researchers trained a language model on a large corpus of chemistry-related text, allowing it to learn the semantic relationships between different chemical ideas and terminology.
They then used this language model to extract relevant chemical concepts that could explain the predictions made by a separate molecular property prediction model. Specifically, they would input a molecule into the property prediction model, get the output prediction, and then use the language model to identify the chemical concepts most closely associated with that prediction.
By connecting the abstract model predictions to interpretable chemical principles, the approach aims to make the predictions more explainable and trustworthy. This could be especially important for safety-critical applications, where being able to understand and validate the model's reasoning is crucial.
The paper demonstrates the effectiveness of this approach through experiments on several molecular property prediction tasks, showing that it can provide meaningful chemical explanations for the model's outputs. The authors also discuss potential limitations and areas for future research in this domain.
Critical Analysis
The key strength of this work is its attempt to bridge the gap between machine learning models and human understanding of chemistry. Providing explainable AI is an important challenge, and this paper offers a novel approach leveraging language models to achieve that for molecular property prediction.
However, the paper does not deeply address some potential limitations. For example, the reliance on language models means the approach may struggle with more complex or esoteric chemical concepts that are not well-captured in the training text. There may also be challenges in scaling the method to handle the full diversity of molecular structures and properties.
Additionally, while the paper demonstrates the method's effectiveness, it does not provide a thorough error analysis or exploration of failure cases. Understanding the model's limitations and blind spots would be important for deploying such a system in real-world, safety-critical applications.
Overall, this is a promising direction for making molecular property prediction models more interpretable and aligned with human chemical reasoning. However, further research is needed to fully address the challenges of providing ,[object Object], for these types of AI systems.
Conclusion
This paper presents an innovative approach to making molecular property prediction models more explainable by aligning their outputs with underlying chemical concepts. By leveraging language models trained on chemistry knowledge, the method can provide interpretable explanations for the model's predictions.
While the work shows promise, there are also important limitations and open questions that merit further exploration. Addressing the scalability and robustness of such explainable AI systems will be crucial for their safe and effective deployment in real-world chemical research and development.
Overall, this research represents an important step towards building more transparent and trustworthy machine learning models for critical scientific applications.
This summary was produced with help from an AI and may contain inaccuracies - check out the links to read the original source documents!
Related Papers
0
Explainable Molecular Property Prediction: Aligning Chemical Concepts with Predictions via Language Models
Zhenzhong Wang, Zehui Lin, Wanyu Lin, Ming Yang, Minggang Zeng, Kay Chen Tan
Providing explainable molecule property predictions is critical for many scientific domains, such as drug discovery and material science. Though transformer-based language models have shown great potential in accurate molecular property prediction, they neither provide chemically meaningful explanations nor faithfully reveal the molecular structure-property relationships. In this work, we develop a new framework for explainable molecular property prediction based on language models, dubbed as Lamole, which can provide chemical concepts-aligned explanations. We first leverage a designated molecular representation -- the Group SELFIES -- as it can provide chemically meaningful semantics. Because attention mechanisms in Transformers can inherently capture relationships within the input, we further incorporate the attention weights and gradients together to generate explanations for capturing the functional group interactions. We then carefully craft a marginal loss to explicitly optimize the explanations to be able to align with the chemists' annotations. We bridge the manifold hypothesis with the elaborated marginal loss to prove that the loss can align the explanations with the tangent space of the data manifold, leading to concept-aligned explanations. Experimental results over six mutagenicity datasets and one hepatotoxicity dataset demonstrate Lamole can achieve comparable classification accuracy and boost the explanation accuracy by up to 14.8%, being the state-of-the-art in explainable molecular property prediction.
Read more6/4/2024

0
Cross-Modal Learning for Chemistry Property Prediction: Large Language Models Meet Graph Machine Learning
Sakhinana Sagar Srinivas, Venkataramana Runkana
In the field of chemistry, the objective is to create novel molecules with desired properties, facilitating accurate property predictions for applications such as material design and drug screening. However, existing graph deep learning methods face limitations that curb their expressive power. To address this, we explore the integration of vast molecular domain knowledge from Large Language Models (LLMs) with the complementary strengths of Graph Neural Networks (GNNs) to enhance performance in property prediction tasks. We introduce a Multi-Modal Fusion (MMF) framework that synergistically harnesses the analytical prowess of GNNs and the linguistic generative and predictive abilities of LLMs, thereby improving accuracy and robustness in predicting molecular properties. Our framework combines the effectiveness of GNNs in modeling graph-structured data with the zero-shot and few-shot learning capabilities of LLMs, enabling improved predictions while reducing the risk of overfitting. Furthermore, our approach effectively addresses distributional shifts, a common challenge in real-world applications, and showcases the efficacy of learning cross-modal representations, surpassing state-of-the-art baselines on benchmark datasets for property prediction tasks.
Read more8/28/2024

0
Molecular Graph Representation Learning Integrating Large Language Models with Domain-specific Small Models
Tianyu Zhang, Yuxiang Ren, Chengbin Hou, Hairong Lv, Xuegong Zhang
Molecular property prediction is a crucial foundation for drug discovery. In recent years, pre-trained deep learning models have been widely applied to this task. Some approaches that incorporate prior biological domain knowledge into the pre-training framework have achieved impressive results. However, these methods heavily rely on biochemical experts, and retrieving and summarizing vast amounts of domain knowledge literature is both time-consuming and expensive. Large Language Models (LLMs) have demonstrated remarkable performance in understanding and efficiently providing general knowledge. Nevertheless, they occasionally exhibit hallucinations and lack precision in generating domain-specific knowledge. Conversely, Domain-specific Small Models (DSMs) possess rich domain knowledge and can accurately calculate molecular domain-related metrics. However, due to their limited model size and singular functionality, they lack the breadth of knowledge necessary for comprehensive representation learning. To leverage the advantages of both approaches in molecular property prediction, we propose a novel Molecular Graph representation learning framework that integrates Large language models and Domain-specific small models (MolGraph-LarDo). Technically, we design a two-stage prompt strategy where DSMs are introduced to calibrate the knowledge provided by LLMs, enhancing the accuracy of domain-specific information and thus enabling LLMs to generate more precise textual descriptions for molecular samples. Subsequently, we employ a multi-modal alignment method to coordinate various modalities, including molecular graphs and their corresponding descriptive texts, to guide the pre-training of molecular representations. Extensive experiments demonstrate the effectiveness of the proposed method.
Read more8/20/2024
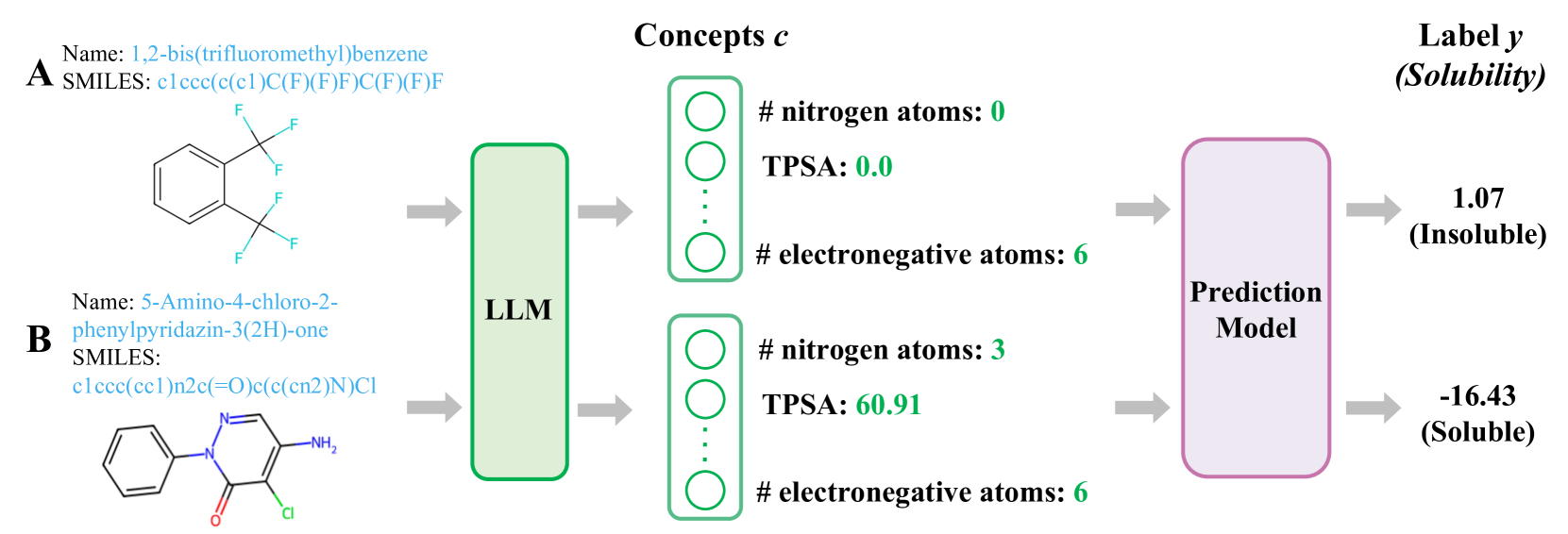
0
Automated Molecular Concept Generation and Labeling with Large Language Models
Shichang Zhang, Botao Xia, Zimin Zhang, Qianli Wu, Fang Sun, Ziniu Hu, Yizhou Sun
Artificial intelligence (AI) is significantly transforming scientific research. Explainable AI methods, such as concept-based models (CMs), are promising for driving new scientific discoveries because they make predictions based on meaningful concepts and offer insights into the prediction process. In molecular science, however, explainable CMs are not as common compared to black-box models like Graph Neural Networks (GNNs), primarily due to their requirement for predefined concepts and manual label for each instance, which demand domain knowledge and can be labor-intensive. This paper introduces a novel framework for Automated Molecular Concept (AutoMolCo) generation and labeling. AutoMolCo leverages the knowledge in Large Language Models (LLMs) to automatically generate predictive molecular concepts and label them for each molecule. Such procedures are repeated through iterative interactions with LLMs to refine concepts, enabling simple linear models on the refined concepts to outperform GNNs and LLM in-context learning on several benchmarks. The whole AutoMolCo framework is automated without any human knowledge inputs in either concept generation, labeling, or refinement, thereby surpassing the limitations of extant CMs while maintaining their explainability and allowing easy intervention. Through systematic experiments on MoleculeNet and High-Throughput Experimentation (HTE) datasets, we demonstrate that the AutoMolCo-induced explainable CMs are beneficial and promising for molecular science research.
Read more6/17/2024