Small Molecule Optimization with Large Language Models

0

Sign in to get full access
Overview
- Explores the use of large language models (LLMs) for optimizing small molecules
- Demonstrates the effectiveness of LLMs in tasks like de novo molecular design and property prediction
- Highlights the potential of LLMs to accelerate drug discovery and chemical research
Plain English Explanation
Large language models (LLMs) are powerful AI systems that can understand and generate human-like text. Researchers have found that these models can also be applied to problems in chemistry and drug discovery.
In this paper, the authors show how LLMs can be used to optimize the properties of small molecules, which are the building blocks of many drugs and other chemicals. They demonstrate that LLMs can generate new molecule designs and accurately predict the properties of these molecules, such as their effectiveness and safety.
The key idea is to treat molecules as a form of "text" that the LLM can understand and manipulate. By training the LLM on a large dataset of existing molecules and their properties, the researchers were able to develop a system that can create new molecules with desired characteristics.
This approach has the potential to greatly accelerate the drug discovery process, as researchers can use LLMs to quickly explore a vast space of possible molecules instead of relying on manual experimentation. The authors also show how LLMs can be used to optimize molecules for specific applications, like making them more potent or less toxic.
Technical Explanation
The researchers used a large language model trained on chemical data to perform various small molecule optimization tasks. They framed these tasks as text generation problems, where the LLM generates SMILES strings (a standard way of representing molecular structures) corresponding to optimized molecules.
For de novo molecular design, the LLM was trained to generate SMILES strings for new molecules with desirable properties, such as high potency and low toxicity. The researchers also showed how the LLM could be used for property prediction, where it learns to estimate the properties of molecules based on their SMILES representations.
Additionally, the authors explored techniques for interpreting and explaining the decisions made by the LLM, allowing for better understanding of how the model is optimizing the molecules.
Critical Analysis
The paper demonstrates the impressive capabilities of LLMs in the domain of small molecule optimization, but it also highlights some important caveats and areas for further research:
- The authors note that the performance of the LLM-based approach is still limited by the quality and breadth of the training data. Expanding the dataset of molecules and their properties could further improve the model's effectiveness.
- There are concerns about the interpretability and "black-box" nature of LLMs, which make it difficult to fully understand the reasoning behind the model's decisions. The authors' work on interpretability techniques is a step in the right direction, but more research is needed in this area.
- The paper focuses on relatively simple molecular optimization tasks. Applying LLMs to more complex problems, such as multi-objective optimization or the design of novel drug scaffolds, could uncover additional challenges and opportunities.
Overall, this research represents a promising step forward in the use of LLMs for accelerating chemical research and drug discovery. However, continued development and careful evaluation will be necessary to fully realize the potential of this approach.
Conclusion
This paper demonstrates the impressive capabilities of large language models (LLMs) in the domain of small molecule optimization. By treating molecules as a form of "text" that the LLM can understand and manipulate, the researchers were able to develop a system that can generate new molecule designs and accurately predict their properties.
The potential implications of this work are significant, as it could greatly accelerate the drug discovery process and enable researchers to explore a much larger space of possible molecules. While the paper highlights some important caveats and areas for further research, it represents a promising step forward in the use of LLMs for advancing chemical science and engineering.
This summary was produced with help from an AI and may contain inaccuracies - check out the links to read the original source documents!
Related Papers
0
Small Molecule Optimization with Large Language Models
Philipp Guevorguian, Menua Bedrosian, Tigran Fahradyan, Gayane Chilingaryan, Hrant Khachatrian, Armen Aghajanyan
Recent advancements in large language models have opened new possibilities for generative molecular drug design. We present Chemlactica and Chemma, two language models fine-tuned on a novel corpus of 110M molecules with computed properties, totaling 40B tokens. These models demonstrate strong performance in generating molecules with specified properties and predicting new molecular characteristics from limited samples. We introduce a novel optimization algorithm that leverages our language models to optimize molecules for arbitrary properties given limited access to a black box oracle. Our approach combines ideas from genetic algorithms, rejection sampling, and prompt optimization. It achieves state-of-the-art performance on multiple molecular optimization benchmarks, including an 8% improvement on Practical Molecular Optimization compared to previous methods. We publicly release the training corpus, the language models and the optimization algorithm.
Read more7/29/2024
0
DrugLLM: Open Large Language Model for Few-shot Molecule Generation
Xianggen Liu, Yan Guo, Haoran Li, Jin Liu, Shudong Huang, Bowen Ke, Jiancheng Lv
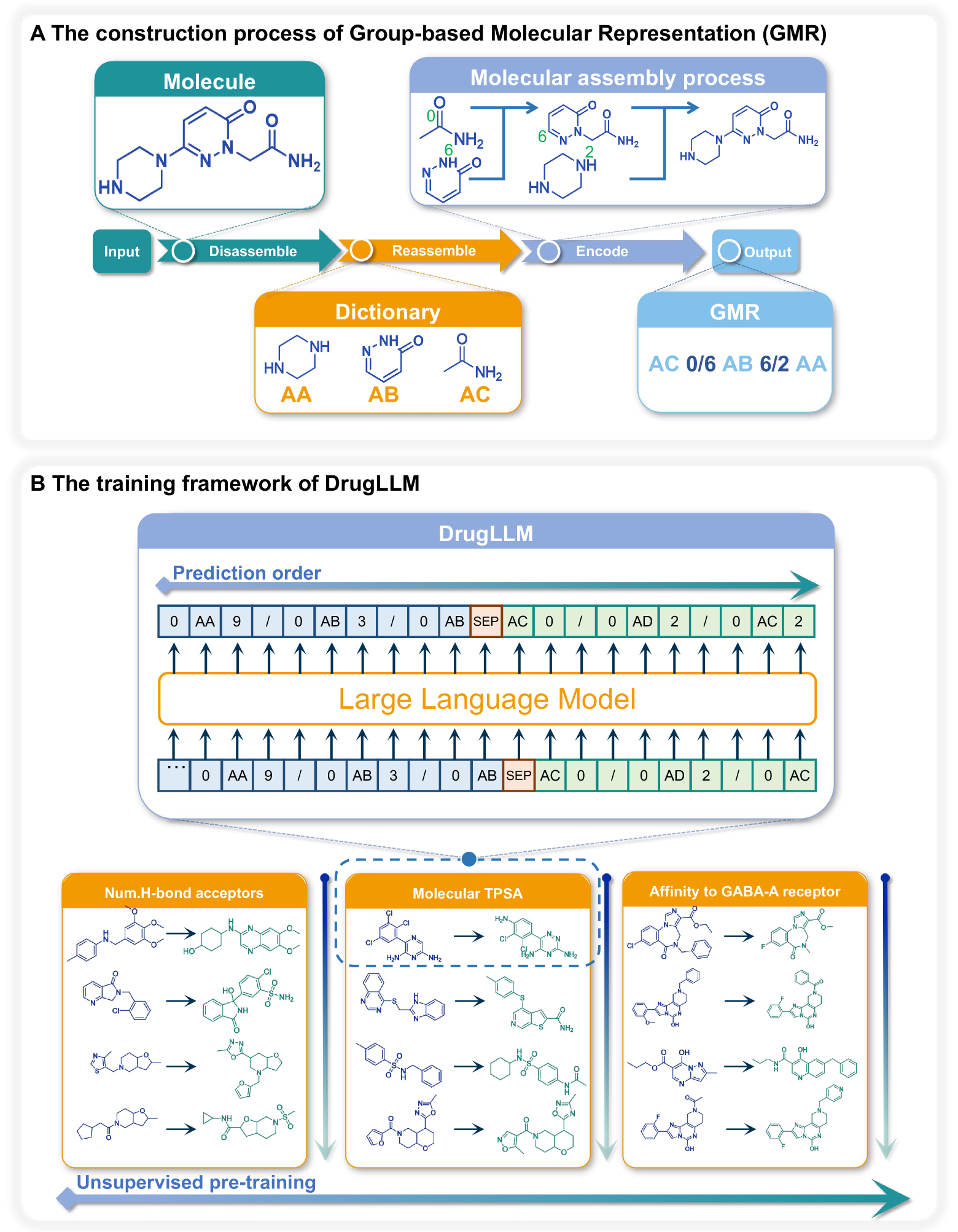
Large Language Models (LLMs) have made great strides in areas such as language processing and computer vision. Despite the emergence of diverse techniques to improve few-shot learning capacity, current LLMs fall short in handling the languages in biology and chemistry. For example, they are struggling to capture the relationship between molecule structure and pharmacochemical properties. Consequently, the few-shot learning capacity of small-molecule drug modification remains impeded. In this work, we introduced DrugLLM, a LLM tailored for drug design. During the training process, we employed Group-based Molecular Representation (GMR) to represent molecules, arranging them in sequences that reflect modifications aimed at enhancing specific molecular properties. DrugLLM learns how to modify molecules in drug discovery by predicting the next molecule based on past modifications. Extensive computational experiments demonstrate that DrugLLM can generate new molecules with expected properties based on limited examples, presenting a powerful few-shot molecule generation capacity.
Read more5/14/2024
💬
0
Token-Mol 1.0: Tokenized drug design with large language model
Jike Wang, Rui Qin, Mingyang Wang, Meijing Fang, Yangyang Zhang, Yuchen Zhu, Qun Su, Qiaolin Gou, Chao Shen, Odin Zhang, Zhenxing Wu, Dejun Jiang, Xujun Zhang, Huifeng Zhao, Xiaozhe Wan, Zhourui Wu, Liwei Liu, Yu Kang, Chang-Yu Hsieh, Tingjun Hou
Significant interests have recently risen in leveraging sequence-based large language models (LLMs) for drug design. However, most current applications of LLMs in drug discovery lack the ability to comprehend three-dimensional (3D) structures, thereby limiting their effectiveness in tasks that explicitly involve molecular conformations. In this study, we introduced Token-Mol, a token-only 3D drug design model. This model encodes all molecular information, including 2D and 3D structures, as well as molecular property data, into tokens, which transforms classification and regression tasks in drug discovery into probabilistic prediction problems, thereby enabling learning through a unified paradigm. Token-Mol is built on the transformer decoder architecture and trained using random causal masking techniques. Additionally, we proposed the Gaussian cross-entropy (GCE) loss function to overcome the challenges in regression tasks, significantly enhancing the capacity of LLMs to learn continuous numerical values. Through a combination of fine-tuning and reinforcement learning (RL), Token-Mol achieves performance comparable to or surpassing existing task-specific methods across various downstream tasks, including pocket-based molecular generation, conformation generation, and molecular property prediction. Compared to existing molecular pre-trained models, Token-Mol exhibits superior proficiency in handling a wider range of downstream tasks essential for drug design. Notably, our approach improves regression task accuracy by approximately 30% compared to similar token-only methods. Token-Mol overcomes the precision limitations of token-only models and has the potential to integrate seamlessly with general models such as ChatGPT, paving the way for the development of a universal artificial intelligence drug design model that facilitates rapid and high-quality drug design by experts.
Read more8/20/2024

0
Crossing New Frontiers: Knowledge-Augmented Large Language Model Prompting for Zero-Shot Text-Based De Novo Molecule Design
Sakhinana Sagar Srinivas, Venkataramana Runkana
Molecule design is a multifaceted approach that leverages computational methods and experiments to optimize molecular properties, fast-tracking new drug discoveries, innovative material development, and more efficient chemical processes. Recently, text-based molecule design has emerged, inspired by next-generation AI tasks analogous to foundational vision-language models. Our study explores the use of knowledge-augmented prompting of large language models (LLMs) for the zero-shot text-conditional de novo molecular generation task. Our approach uses task-specific instructions and a few demonstrations to address distributional shift challenges when constructing augmented prompts for querying LLMs to generate molecules consistent with technical descriptions. Our framework proves effective, outperforming state-of-the-art (SOTA) baseline models on benchmark datasets.
Read more8/23/2024