DrugLLM: Open Large Language Model for Few-shot Molecule Generation

0
Sign in to get full access
Overview
- This paper introduces DrugLLM, an open-source large language model (LLM) trained specifically for few-shot molecule generation.
- The model is designed to generate novel drug-like molecules by learning from a small number of examples, rather than requiring large datasets for training.
- The paper demonstrates DrugLLM's strong performance on various molecule generation and property prediction tasks, showcasing its potential to accelerate drug discovery.
Plain English Explanation
DrugLLM: Open Large Language Model for Few-shot Molecule Generation is a research paper that describes a new artificial intelligence (AI) model called DrugLLM. This model is a type of large language model (LLM), which is a powerful AI system that can understand and generate human-like text.
The key feature of DrugLLM is that it is specifically designed to work with molecules, the building blocks of chemicals and drugs. Unlike many other AI models that require large datasets to learn, DrugLLM can generate new drug-like molecules by learning from just a few examples. This "few-shot" learning capability is important because it can help speed up the process of discovering new drugs, which is typically very time-consuming and expensive.
The paper shows that DrugLLM performs well on a variety of tasks related to molecules, such as generating new molecules and predicting their properties. This suggests that DrugLLM could be a valuable tool for researchers and companies working on developing new drugs and other chemical compounds.
Technical Explanation
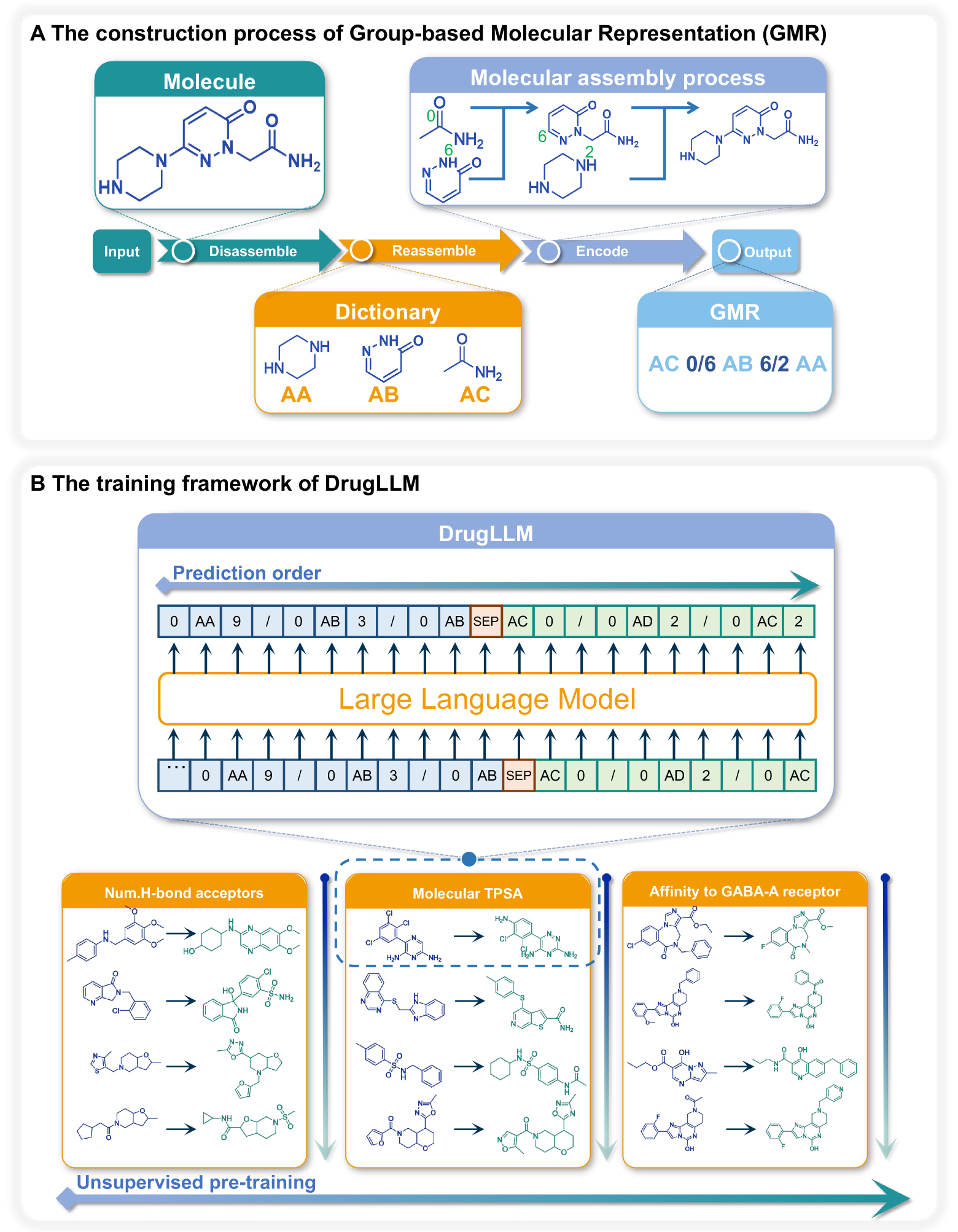
DrugLLM: Open Large Language Model for Few-shot Molecule Generation presents a novel large language model (LLM) called DrugLLM that is specifically designed for few-shot molecule generation. The model is trained on a large corpus of chemical data, including molecular structures, properties, and reactions, and is able to generate new drug-like molecules by learning from just a few examples.
The authors describe the architecture of DrugLLM, which is based on the Transformer language model. They also detail the training process, which involves pretraining the model on a diverse set of chemical data, followed by fine-tuning on specific molecule generation tasks.
To evaluate the performance of DrugLLM, the researchers conducted a series of experiments on various molecule generation and property prediction benchmarks. The results show that DrugLLM outperforms previous state-of-the-art models, demonstrating its strong few-shot learning capabilities and its potential to accelerate drug discovery.
The paper also compares DrugLLM to other LLMs for chemistry, assessing its capabilities as a "superhuman chemist", and exploring its ability to learn the context of molecules.
Critical Analysis
The paper presents a compelling case for the potential of DrugLLM to accelerate drug discovery, but it also acknowledges several limitations and areas for further research.
One key caveat is that the model's performance is still dependent on the quality and diversity of the training data, and the authors note that expanding the dataset could further improve the model's capabilities. Additionally, the paper does not address potential biases or safety concerns that may arise from using such a powerful AI system for drug development.
Another area for further exploration is the interpretability of DrugLLM's decision-making process. While the model demonstrates strong performance, it remains a "black box" in terms of understanding why it generates certain molecules or makes specific predictions. Developing more transparent and explainable AI systems could be crucial for building trust and ensuring the responsible use of these technologies in the pharmaceutical industry.
Despite these limitations, the framework for multimodal learning proposed in the paper suggests that DrugLLM and similar LLMs could have far-reaching implications for the future of drug discovery and chemical research.
Conclusion
The DrugLLM paper presents a significant advancement in the field of molecule generation by introducing an open-source large language model that can generate novel drug-like compounds from just a few examples. The model's strong performance on a range of benchmarks highlights its potential to accelerate the drug discovery process and revolutionize how researchers approach the development of new chemicals and pharmaceuticals.
While the paper acknowledges several areas for further research and improvement, the overall impact of DrugLLM and similar LLMs for chemistry could be profound. As these technologies continue to evolve, they may unlock new avenues for scientific discovery and lead to the development of life-saving medications and treatments.
This summary was produced with help from an AI and may contain inaccuracies - check out the links to read the original source documents!
Related Papers
0
DrugLLM: Open Large Language Model for Few-shot Molecule Generation
Xianggen Liu, Yan Guo, Haoran Li, Jin Liu, Shudong Huang, Bowen Ke, Jiancheng Lv
Large Language Models (LLMs) have made great strides in areas such as language processing and computer vision. Despite the emergence of diverse techniques to improve few-shot learning capacity, current LLMs fall short in handling the languages in biology and chemistry. For example, they are struggling to capture the relationship between molecule structure and pharmacochemical properties. Consequently, the few-shot learning capacity of small-molecule drug modification remains impeded. In this work, we introduced DrugLLM, a LLM tailored for drug design. During the training process, we employed Group-based Molecular Representation (GMR) to represent molecules, arranging them in sequences that reflect modifications aimed at enhancing specific molecular properties. DrugLLM learns how to modify molecules in drug discovery by predicting the next molecule based on past modifications. Extensive computational experiments demonstrate that DrugLLM can generate new molecules with expected properties based on limited examples, presenting a powerful few-shot molecule generation capacity.
Read more5/14/2024

0
MolecularGPT: Open Large Language Model (LLM) for Few-Shot Molecular Property Prediction
Yuyan Liu, Sirui Ding, Sheng Zhou, Wenqi Fan, Qiaoyu Tan
Molecular property prediction (MPP) is a fundamental and crucial task in drug discovery. However, prior methods are limited by the requirement for a large number of labeled molecules and their restricted ability to generalize for unseen and new tasks, both of which are essential for real-world applications. To address these challenges, we present MolecularGPT for few-shot MPP. From a perspective on instruction tuning, we fine-tune large language models (LLMs) based on curated molecular instructions spanning over 1000 property prediction tasks. This enables building a versatile and specialized LLM that can be adapted to novel MPP tasks without any fine-tuning through zero- and few-shot in-context learning (ICL). MolecularGPT exhibits competitive in-context reasoning capabilities across 10 downstream evaluation datasets, setting new benchmarks for few-shot molecular prediction tasks. More importantly, with just two-shot examples, MolecularGPT can outperform standard supervised graph neural network methods on 4 out of 7 datasets. It also excels state-of-the-art LLM baselines by up to 16.6% increase on classification accuracy and decrease of 199.17 on regression metrics (e.g., RMSE) under zero-shot. This study demonstrates the potential of LLMs as effective few-shot molecular property predictors. The code is available at https://github.com/NYUSHCS/MolecularGPT.
Read more6/21/2024

0
Small Molecule Optimization with Large Language Models
Philipp Guevorguian, Menua Bedrosian, Tigran Fahradyan, Gayane Chilingaryan, Hrant Khachatrian, Armen Aghajanyan
Recent advancements in large language models have opened new possibilities for generative molecular drug design. We present Chemlactica and Chemma, two language models fine-tuned on a novel corpus of 110M molecules with computed properties, totaling 40B tokens. These models demonstrate strong performance in generating molecules with specified properties and predicting new molecular characteristics from limited samples. We introduce a novel optimization algorithm that leverages our language models to optimize molecules for arbitrary properties given limited access to a black box oracle. Our approach combines ideas from genetic algorithms, rejection sampling, and prompt optimization. It achieves state-of-the-art performance on multiple molecular optimization benchmarks, including an 8% improvement on Practical Molecular Optimization compared to previous methods. We publicly release the training corpus, the language models and the optimization algorithm.
Read more7/29/2024
💬
0
ChemLLM: A Chemical Large Language Model
Di Zhang, Wei Liu, Qian Tan, Jingdan Chen, Hang Yan, Yuliang Yan, Jiatong Li, Weiran Huang, Xiangyu Yue, Wanli Ouyang, Dongzhan Zhou, Shufei Zhang, Mao Su, Han-Sen Zhong, Yuqiang Li
Large language models (LLMs) have made impressive progress in chemistry applications. However, the community lacks an LLM specifically designed for chemistry. The main challenges are two-fold: firstly, most chemical data and scientific knowledge are stored in structured databases, which limits the model's ability to sustain coherent dialogue when used directly. Secondly, there is an absence of objective and fair benchmark that encompass most chemistry tasks. Here, we introduce ChemLLM, a comprehensive framework that features the first LLM dedicated to chemistry. It also includes ChemData, a dataset specifically designed for instruction tuning, and ChemBench, a robust benchmark covering nine essential chemistry tasks. ChemLLM is adept at performing various tasks across chemical disciplines with fluid dialogue interaction. Notably, ChemLLM achieves results comparable to GPT-4 on the core chemical tasks and demonstrates competitive performance with LLMs of similar size in general scenarios. ChemLLM paves a new path for exploration in chemical studies, and our method of incorporating structured chemical knowledge into dialogue systems sets a new standard for developing LLMs in various scientific fields. Codes, Datasets, and Model weights are publicly accessible at https://hf.co/AI4Chem
Read more4/26/2024