Transferable Neural Wavefunctions for Solids

0
Sign in to get full access
Overview
- This paper presents a novel approach for modeling quantum mechanical wavefunctions in solid materials using transferable neural networks.
- The researchers demonstrate the effectiveness of their neural wavefunction models on 1D hydrogen chain systems and 3D crystalline solids.
- The proposed models show promising results in accurately capturing the electronic structure of materials and could have significant implications for materials discovery and design.
Plain English Explanation
The paper explores a new way to model the behavior of electrons in solid materials using advanced artificial intelligence (AI) techniques. Electrons in solids exhibit complex quantum mechanical behavior that is crucial for understanding and predicting the properties of materials. However, modeling these quantum effects accurately is extremely challenging.
The researchers developed a special type of neural network model that can learn to represent the quantum mechanical wavefunctions of electrons in solid materials. These neural wavefunctions are "transferable," meaning they can be applied to study different materials beyond just the ones used in the training process. This is an important capability, as it allows the models to be used for discovering and designing new materials with desired properties.
The team tested their neural wavefunction models on simple 1D chains of hydrogen atoms as well as more complex 3D crystalline solids. The results showed that the models could accurately capture the quantum mechanical behavior of the electrons in these systems, suggesting they may be useful for studying a wide range of solid materials.
Overall, this work represents an exciting advance in the field of computational materials science, as it provides a powerful new tool for modeling the fundamental quantum properties of materials. By leveraging the flexibility and learning capabilities of neural networks, the researchers have developed an approach that could accelerate the discovery and development of new materials with important applications in technology, energy, and beyond.
Technical Explanation
The paper introduces a novel approach for modeling the quantum mechanical wavefunctions of electrons in solid materials using transferable neural networks. The researchers developed a neural network architecture that can learn to represent the complex, many-body wavefunctions of electrons in solids, which are governed by the fundamental laws of quantum mechanics.
The key innovation of this work is the ability to train these neural wavefunction models on a limited set of materials and then apply them to study the electronic structure of other, previously unseen materials. This "transferability" is achieved by incorporating physical constraints and symmetries into the neural network design, which allows the models to generalize beyond the specific training data.
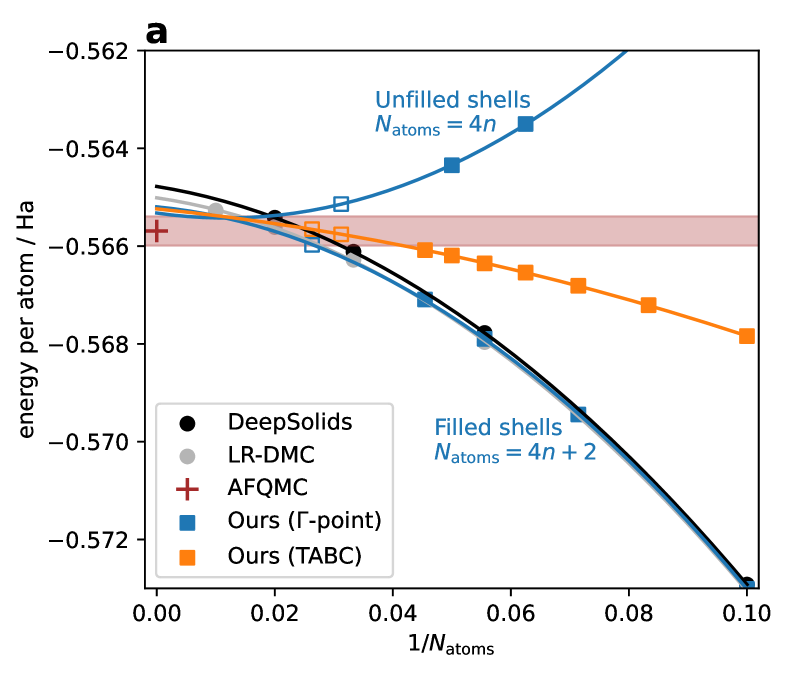
The team evaluated their neural wavefunction models on two types of solid systems: 1D hydrogen chains and 3D crystalline solids. For the 1D hydrogen chains, the neural models were able to accurately capture the ground state and excited state wavefunctions, as well as the total energy of the system. The researchers then demonstrated the transferability of the models by applying them to study 3D crystalline solids, including silicon and magnesium oxide, and showed that the neural wavefunctions could faithfully reproduce the electronic structure of these materials.
The success of the neural wavefunction models on these diverse solid systems suggests that the approach could be a powerful tool for materials discovery and design. By leveraging the flexibility and learning capabilities of neural networks, the researchers have developed a framework that can capture the complex quantum mechanical behavior of electrons in solids and potentially accelerate the discovery of new materials with desirable properties.
Critical Analysis
The paper presents a compelling approach for modeling the quantum mechanical wavefunctions of electrons in solid materials using transferable neural networks. The results demonstrate the ability of the models to accurately capture the electronic structure of both simple 1D hydrogen chains and more complex 3D crystalline solids, which is a significant achievement.
One potential limitation of the work, as acknowledged by the authors, is the relatively small size of the training datasets used for the 3D crystalline solids. Expanding the training data to include a broader range of materials could further improve the generalization and transferability of the neural wavefunction models. Additionally, the paper does not explore the computational efficiency and scalability of the approach, which would be important considerations for applying the models to large-scale materials discovery and optimization tasks.
Another area for further research could be the integration of the neural wavefunction models with other AI-driven materials discovery techniques, such as generative models or reinforcement learning. By combining the strengths of different AI approaches, researchers may be able to develop even more powerful and comprehensive tools for computational materials science.
Overall, this paper represents an important step forward in the application of neural networks to the modeling of quantum mechanical phenomena in solids. The transferable neural wavefunction models demonstrated in this work have the potential to significantly impact materials discovery and design, and the continued development of this line of research could yield transformative insights and advancements in the field of computational materials science.
Conclusion
This paper presents a novel approach for modeling the quantum mechanical wavefunctions of electrons in solid materials using transferable neural networks. The researchers have developed a flexible neural network architecture that can learn to represent the complex, many-body behavior of electrons in solids and then apply these models to study the electronic structure of a wide range of materials.
The demonstrated results on both 1D hydrogen chains and 3D crystalline solids suggest that the neural wavefunction models can accurately capture the quantum mechanical properties of electrons in these systems. This capability could have significant implications for accelerating the discovery and design of new materials with desirable properties, as the transferability of the models allows them to be applied beyond the specific training data.
Overall, this work represents an important advancement in the field of computational materials science, as it provides a powerful new tool for modeling the fundamental quantum behavior of electrons in solids. By leveraging the flexibility and learning capabilities of neural networks, the researchers have developed an approach that could help unlock new materials with transformative applications in technology, energy, and beyond.
This summary was produced with help from an AI and may contain inaccuracies - check out the links to read the original source documents!
Related Papers
0
Transferable Neural Wavefunctions for Solids
Leon Gerard, Michael Scherbela, Halvard Sutterud, Matthew Foulkes, Philipp Grohs
Deep-Learning-based Variational Monte Carlo (DL-VMC) has recently emerged as a highly accurate approach for finding approximate solutions to the many-electron Schrodinger equation. Despite its favorable scaling with the number of electrons, $mathcal{O}(n_text{el}^{4})$, the practical value of DL-VMC is limited by the high cost of optimizing the neural network weights for every system studied. To mitigate this problem, recent research has proposed optimizing a single neural network across multiple systems, reducing the cost per system. Here we extend this approach to solids, where similar but distinct calculations using different geometries, boundary conditions, and supercell sizes are often required. We show how to optimize a single ansatz across all of these variations, reducing the required number of optimization steps by an order of magnitude. Furthermore, we exploit the transfer capabilities of a pre-trained network. We successfully transfer a network, pre-trained on 2x2x2 supercells of LiH, to 3x3x3 supercells. This reduces the number of optimization steps required to simulate the large system by a factor of 50 compared to previous work.
Read more5/14/2024

0
Highly Accurate Real-space Electron Densities with Neural Networks
Lixue Cheng, P. Bern'at Szab'o, Zeno Schatzle, Derk Kooi, Jonas Kohler, Klaas J. H. Giesbertz, Frank No'e, Jan Hermann, Paola Gori-Giorgi, Adam Foster
Variational ab-initio methods in quantum chemistry stand out among other methods in providing direct access to the wave function. This allows in principle straightforward extraction of any other observable of interest, besides the energy, but in practice this extraction is often technically difficult and computationally impractical. Here, we consider the electron density as a central observable in quantum chemistry and introduce a novel method to obtain accurate densities from real-space many-electron wave functions by representing the density with a neural network that captures known asymptotic properties and is trained from the wave function by score matching and noise-contrastive estimation. We use variational quantum Monte Carlo with deep-learning ansatze (deep QMC) to obtain highly accurate wave functions free of basis set errors, and from them, using our novel method, correspondingly accurate electron densities, which we demonstrate by calculating dipole moments, nuclear forces, contact densities, and other density-based properties.
Read more9/4/2024
🧠
0
Neural Wave Functions for Superfluids
Wan Tong Lou, Halvard Sutterud, Gino Cassella, W. M. C. Foulkes, Johannes Knolle, David Pfau, James S. Spencer
Understanding superfluidity remains a major goal of condensed matter physics. Here we tackle this challenge utilizing the recently developed Fermionic neural network (FermiNet) wave function Ansatz [D. Pfau et al., Phys. Rev. Res. 2, 033429 (2020).] for variational Monte Carlo calculations. We study the unitary Fermi gas, a system with strong, short-range, two-body interactions known to possess a superfluid ground state but difficult to describe quantitatively. We demonstrate key limitations of the FermiNet Ansatz in studying the unitary Fermi gas and propose a simple modification based on the idea of an antisymmetric geminal power singlet (AGPs) wave function. The new AGPs FermiNet outperforms the original FermiNet significantly in paired systems, giving results which are more accurate than fixed-node diffusion Monte Carlo and are consistent with experiment. We prove mathematically that the new Ansatz, which only differs from the original Ansatz by the method of antisymmetrization, is a strict generalization of the original FermiNet architecture, despite the use of fewer parameters. Our approach shares several advantages with the original FermiNet: the use of a neural network removes the need for an underlying basis set; and the flexibility of the network yields extremely accurate results within a variational quantum Monte Carlo framework that provides access to unbiased estimates of arbitrary ground-state expectation values. We discuss how the method can be extended to study other superfluids.
Read more6/11/2024
0
Machine learning approach for vibronically renormalized electronic band structures
Niraj Aryal, Sheng Zhang, Weiguo Yin, Gia-Wei Chern
We present a machine learning (ML) method for efficient computation of vibrational thermal expectation values of physical properties from first principles. Our approach is based on the non-perturbative frozen phonon formulation in which stochastic Monte Carlo algorithm is employed to sample configurations of nuclei in a supercell at finite temperatures based on a first-principles phonon model. A deep-learning neural network is trained to accurately predict physical properties associated with sampled phonon configurations, thus bypassing the time-consuming {em ab initio} calculations. To incorporate the point-group symmetry of the electronic system into the ML model, group-theoretical methods are used to develop a symmetry-invariant descriptor for phonon configurations in the supercell. We apply our ML approach to compute the temperature dependent electronic energy gap of silicon based on density functional theory (DFT). We show that, with less than a hundred DFT calculations for training the neural network model, an order of magnitude larger number of sampling can be achieved for the computation of the vibrational thermal expectation values. Our work highlights the promising potential of ML techniques for finite temperature first-principles electronic structure methods.
Read more9/4/2024