Accurate Spatial Gene Expression Prediction by integrating Multi-resolution features

0

Sign in to get full access
Overview
- This paper presents a novel method for accurately predicting spatial gene expression by integrating multi-resolution features.
- The approach leverages both local and global information to improve the accuracy of gene expression prediction compared to previous methods.
- The authors demonstrate the effectiveness of their approach on several datasets, showing significant performance improvements over state-of-the-art techniques.
Plain English Explanation
Genes are the basic units of heredity that contain the instructions for how our cells function. Each gene has a unique pattern of expression, which means how much that gene is "turned on" and producing its corresponding proteins. Understanding the spatial distribution of gene expression across different regions of a tissue or organ is crucial for studying biological processes and diseases.
The authors of this paper have developed a new method to predict the spatial expression patterns of genes more accurately than previous techniques. Their approach combines information from different scales or "resolutions" of the tissue, including both local details and broader, global features. By integrating these multi-resolution features, the model can better capture the complex spatial relationships and dynamics of gene expression.
The researchers tested their method on several datasets and showed that it outperforms existing state-of-the-art models in predicting spatial gene expression. This improved ability to map gene activity across tissues could lead to better understanding of biological pathways and help identify regions of interest for further study.
Overall, this work represents an important advance in the field of spatial genomics, providing a powerful tool for fusing pathology and genomic data and potentially enabling more accurate interpretation of weakly supervised models in spatial biology.
Technical Explanation
The key innovation of this paper is the integration of multi-resolution features to improve the accuracy of spatial gene expression prediction. The authors propose a deep learning architecture that combines local patch-level information with global context from the entire tissue sample.
Specifically, the model takes in a tissue image and extracts features at multiple scales using a cross-modal diffusion modeling approach. These features capture both fine-grained details and broader spatial relationships. The multi-resolution features are then fused and fed into a prediction head to estimate the expression level of each gene at each spatial location.
The authors evaluate their method on several spatial transcriptomics datasets, including the Visium and Slide-seqV2 datasets. They show that their approach significantly outperforms previous state-of-the-art techniques, such as DestVI and Tangram, in predicting spatial gene expression patterns.
Further analysis reveals that the model's ability to leverage both local and global information is key to its superior performance. The multi-resolution features allow the model to better capture the complex spatial dynamics and underlying biological processes driving gene expression.
Critical Analysis
The authors acknowledge several limitations of their work. First, the method requires a pre-existing tissue image, which may not always be available in real-world applications. They suggest exploring ways to integrate the model with spatial transcriptomics data acquisition pipelines.
Additionally, the current implementation is computationally intensive, as it requires processing the entire tissue image at multiple scales. The authors note that future work could explore more efficient architectures or inference techniques to improve the scalability of the approach.
Another potential issue is the interpretability of the model's predictions. While the multi-resolution features capture meaningful spatial patterns, it may be challenging to directly interpret the biological significance of the model's outputs. Developing methods to spatially interpret the model's decisions could further enhance the model's utility for biological research.
Overall, this paper presents a promising approach to spatial gene expression prediction that leverages the power of multi-resolution features. The demonstrated performance improvements over existing methods suggest that this work could have a substantial impact on the field of spatial genomics. However, the practical deployment and interpretability of the model remain important areas for future research.
Conclusion
This paper introduces a novel method for accurately predicting spatial gene expression patterns by integrating multi-resolution features. The approach combines local and global information to capture the complex spatial dynamics of gene expression, leading to significant performance improvements over state-of-the-art techniques.
The ability to map gene activity across tissues with high accuracy could enable better understanding of biological pathways, identification of regions of interest, and fusion of pathology and genomic data. This work represents an important advance in the field of spatial genomics and could have far-reaching implications for biological research and clinical applications.
This summary was produced with help from an AI and may contain inaccuracies - check out the links to read the original source documents!
Related Papers
0
Accurate Spatial Gene Expression Prediction by integrating Multi-resolution features
Youngmin Chung, Ji Hun Ha, Kyeong Chan Im, Joo Sang Lee
Recent advancements in Spatial Transcriptomics (ST) technology have facilitated detailed gene expression analysis within tissue contexts. However, the high costs and methodological limitations of ST necessitate a more robust predictive model. In response, this paper introduces TRIPLEX, a novel deep learning framework designed to predict spatial gene expression from Whole Slide Images (WSIs). TRIPLEX uniquely harnesses multi-resolution features, capturing cellular morphology at individual spots, the local context around these spots, and the global tissue organization. By integrating these features through an effective fusion strategy, TRIPLEX achieves accurate gene expression prediction. Our comprehensive benchmark study, conducted on three public ST datasets and supplemented with Visium data from 10X Genomics, demonstrates that TRIPLEX outperforms current state-of-the-art models in Mean Squared Error (MSE), Mean Absolute Error (MAE), and Pearson Correlation Coefficient (PCC). The model's predictions align closely with ground truth gene expression profiles and tumor annotations, underscoring TRIPLEX's potential in advancing cancer diagnosis and treatment.
Read more4/26/2024

0
Multimodal contrastive learning for spatial gene expression prediction using histology images
Wenwen Min, Zhiceng Shi, Jun Zhang, Jun Wan, Changmiao Wang
In recent years, the advent of spatial transcriptomics (ST) technology has unlocked unprecedented opportunities for delving into the complexities of gene expression patterns within intricate biological systems. Despite its transformative potential, the prohibitive cost of ST technology remains a significant barrier to its widespread adoption in large-scale studies. An alternative, more cost-effective strategy involves employing artificial intelligence to predict gene expression levels using readily accessible whole-slide images (WSIs) stained with Hematoxylin and Eosin (H&E). However, existing methods have yet to fully capitalize on multimodal information provided by H&E images and ST data with spatial location. In this paper, we propose textbf{mclSTExp}, a multimodal contrastive learning with Transformer and Densenet-121 encoder for Spatial Transcriptomics Expression prediction. We conceptualize each spot as a word, integrating its intrinsic features with spatial context through the self-attention mechanism of a Transformer encoder. This integration is further enriched by incorporating image features via contrastive learning, thereby enhancing the predictive capability of our model. Our extensive evaluation of textbf{mclSTExp} on two breast cancer datasets and a skin squamous cell carcinoma dataset demonstrates its superior performance in predicting spatial gene expression. Moreover, mclSTExp has shown promise in interpreting cancer-specific overexpressed genes, elucidating immune-related genes, and identifying specialized spatial domains annotated by pathologists. Our source code is available at https://github.com/shizhiceng/mclSTExp.
Read more7/12/2024
0
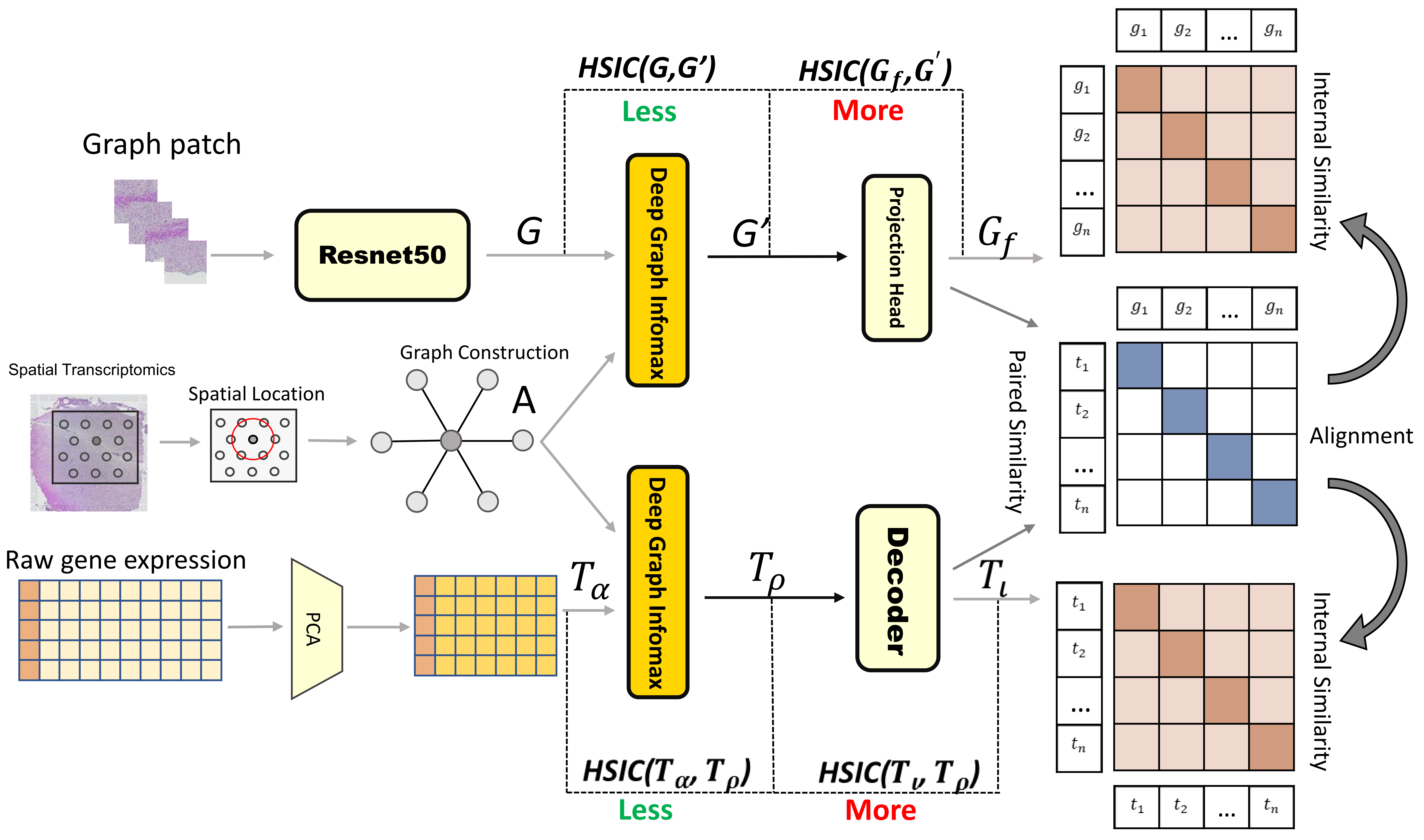
Spatially Resolved Gene Expression Prediction from Histology via Multi-view Graph Contrastive Learning with HSIC-bottleneck Regularization
Changxi Chi, Hang Shi, Qi Zhu, Daoqiang Zhang, Wei Shao
The rapid development of spatial transcriptomics(ST) enables the measurement of gene expression at spatial resolution, making it possible to simultaneously profile the gene expression, spatial locations of spots, and the matched histopathological images. However, the cost for collecting ST data is much higher than acquiring histopathological images, and thus several studies attempt to predict the gene expression on ST by leveraging their corresponding histopathological images. Most of the existing image-based gene prediction models treat the prediction task on each spot of ST data independently, which ignores the spatial dependency among spots. In addition, while the histology images share phenotypic characteristics with the ST data, it is still challenge to extract such common information to help align paired image and expression representations. To address the above issues, we propose a Multi-view Graph Contrastive Learning framework with HSIC-bottleneck Regularization(ST-GCHB) aiming at learning shared representation to help impute the gene expression of the queried imagingspots by considering their spatial dependency.
Read more6/19/2024
0
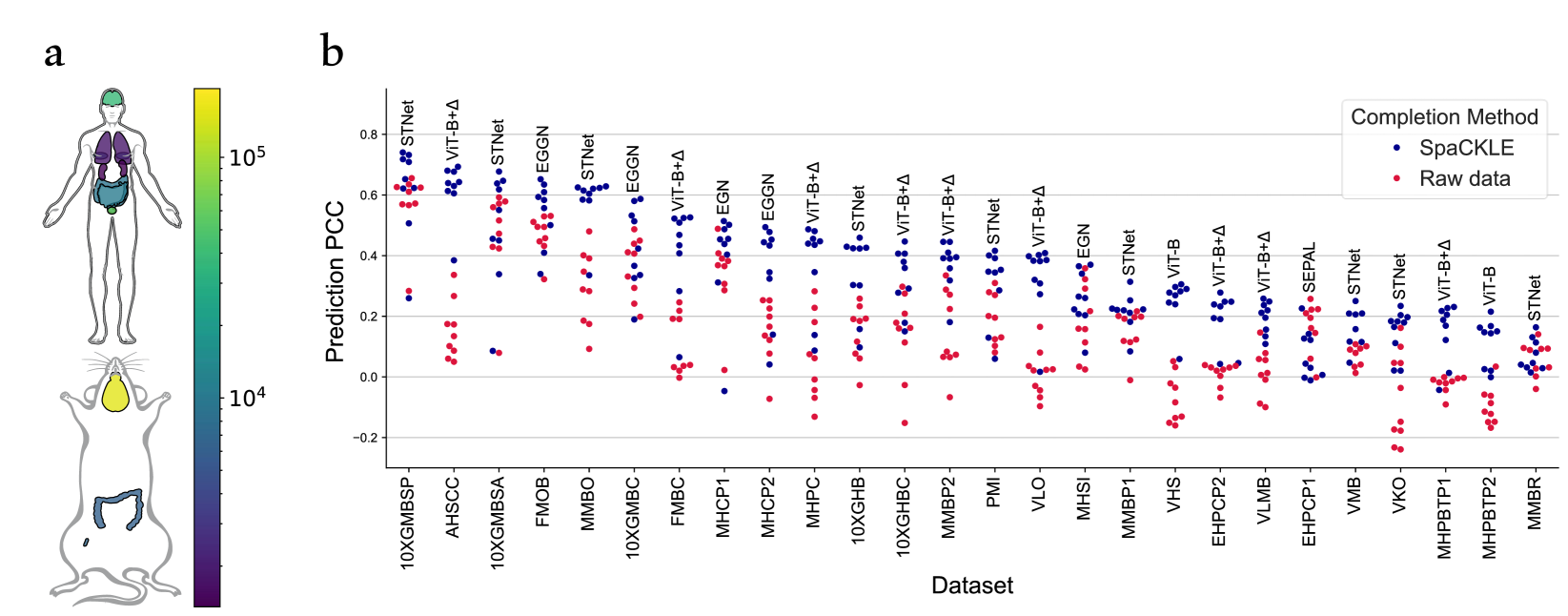
Enhancing Gene Expression Prediction from Histology Images with Spatial Transcriptomics Completion
Gabriel Mejia, Daniela Ruiz, Paula C'ardenas, Leonardo Manrique, Daniela Vega, Pablo Arbel'aez
Spatial Transcriptomics is a novel technology that aligns histology images with spatially resolved gene expression profiles. Although groundbreaking, it struggles with gene capture yielding high corruption in acquired data. Given potential applications, recent efforts have focused on predicting transcriptomic profiles solely from histology images. However, differences in databases, preprocessing techniques, and training hyperparameters hinder a fair comparison between methods. To address these challenges, we present a systematically curated and processed database collected from 26 public sources, representing an 8.6-fold increase compared to previous works. Additionally, we propose a state-of-the-art transformer based completion technique for inferring missing gene expression, which significantly boosts the performance of transcriptomic profile predictions across all datasets. Altogether, our contributions constitute the most comprehensive benchmark of gene expression prediction from histology images to date and a stepping stone for future research on spatial transcriptomics.
Read more7/19/2024