CCPL: Cross-modal Contrastive Protein Learning

0
🖼️
Sign in to get full access
Overview
- Effective protein representation learning is crucial for predicting protein functions
- Traditional methods pretrain protein language models on unlabeled amino acid sequences, then finetune on labeled data
- These methods underutilize the potential of protein structures, which are vital for function determination
- Structural representation techniques rely heavily on annotated data, limiting their generalizability
- Structural pretraining methods can distort actual protein structures
Plain English Explanation
Proteins are the building blocks of life, and understanding how they work is essential for many applications, such as drug discovery. Protein representation learning involves developing mathematical models to capture the key features of proteins that are relevant for predicting their functions.
Traditional methods often start by training a protein language model on a large dataset of protein sequences, without any labels or annotations. This helps the model learn the patterns and grammar of protein sequences. Then, the model is "fine-tuned" on a smaller dataset that has labeled information about the proteins' functions.
While this approach can be effective, it doesn't fully utilize the information contained in protein structures, which are critical for understanding how proteins work. Structural representation techniques that focus on protein structures often rely heavily on annotated data, which can limit their ability to generalize to new proteins.
Additionally, methods that try to explicitly model protein structures during pretraining, similar to how language models are pretrained on natural language, can sometimes distort the actual structures of the proteins.
Technical Explanation
In this work, the researchers introduce a novel unsupervised protein structure representation pretraining method called Cross-Modal Contrastive Protein Learning (CCPL). CCPL leverages a robust protein language model and uses unsupervised contrastive alignment to enhance structure learning, incorporating self-supervised structural constraints to maintain the intrinsic structural information of the proteins.
The key idea behind CCPL is to jointly learn representations from both the protein sequence and structure in an unsupervised manner, without relying on annotated data. This allows the model to capture the rich structural information of proteins while still benefiting from the patterns learned in the sequence domain.
The researchers evaluate their CCPL model on various benchmarks and demonstrate that it outperforms other state-of-the-art methods in tasks related to protein function prediction and structure-based analysis.
Critical Analysis
The researchers acknowledge that their method, like others, has some limitations. For example, the self-supervised structural constraints used in CCPL may not fully capture all the nuances of protein structure, and there may be room for further improvement in the unsupervised alignment of sequence and structure representations.
Additionally, the paper does not provide a detailed analysis of the computational cost and training time required for CCPL, which could be an important consideration for real-world applications.
Overall, the CCPL method represents an important step forward in the field of protein representation learning, as it demonstrates the potential benefits of jointly learning from both sequence and structure information in an unsupervised manner. Further research and refinement of this approach could lead to even more powerful tools for understanding and predicting protein function.
Conclusion
The paper introduces a novel unsupervised protein structure representation pretraining method called CCPL, which leverages a robust protein language model and uses unsupervised contrastive alignment to enhance structure learning. CCPL incorporates self-supervised structural constraints to maintain the intrinsic structural information of proteins, allowing it to outperform other state-of-the-art methods on various benchmarks.
This work highlights the importance of jointly learning from both protein sequence and structure information, and demonstrates the potential of unsupervised approaches to overcome the limitations of methods that rely heavily on annotated data. As the field of protein representation learning continues to evolve, techniques like CCPL may play an increasingly important role in advancing our understanding of protein function and supporting a wide range of applications, from drug discovery to biotechnology.
This summary was produced with help from an AI and may contain inaccuracies - check out the links to read the original source documents!
Related Papers
🖼️
0
CCPL: Cross-modal Contrastive Protein Learning
Jiangbin Zheng, Stan Z. Li
Effective protein representation learning is crucial for predicting protein functions. Traditional methods often pretrain protein language models on large, unlabeled amino acid sequences, followed by finetuning on labeled data. While effective, these methods underutilize the potential of protein structures, which are vital for function determination. Common structural representation techniques rely heavily on annotated data, limiting their generalizability. Moreover, structural pretraining methods, similar to natural language pretraining, can distort actual protein structures. In this work, we introduce a novel unsupervised protein structure representation pretraining method, cross-modal contrastive protein learning (CCPL). CCPL leverages a robust protein language model and uses unsupervised contrastive alignment to enhance structure learning, incorporating self-supervised structural constraints to maintain intrinsic structural information. We evaluated our model across various benchmarks, demonstrating the framework's superiority.
Read more9/5/2024
0
Learning the Language of Protein Structure
Benoit Gaujac, J'er'emie Don`a, Liviu Copoiu, Timothy Atkinson, Thomas Pierrot, Thomas D. Barrett
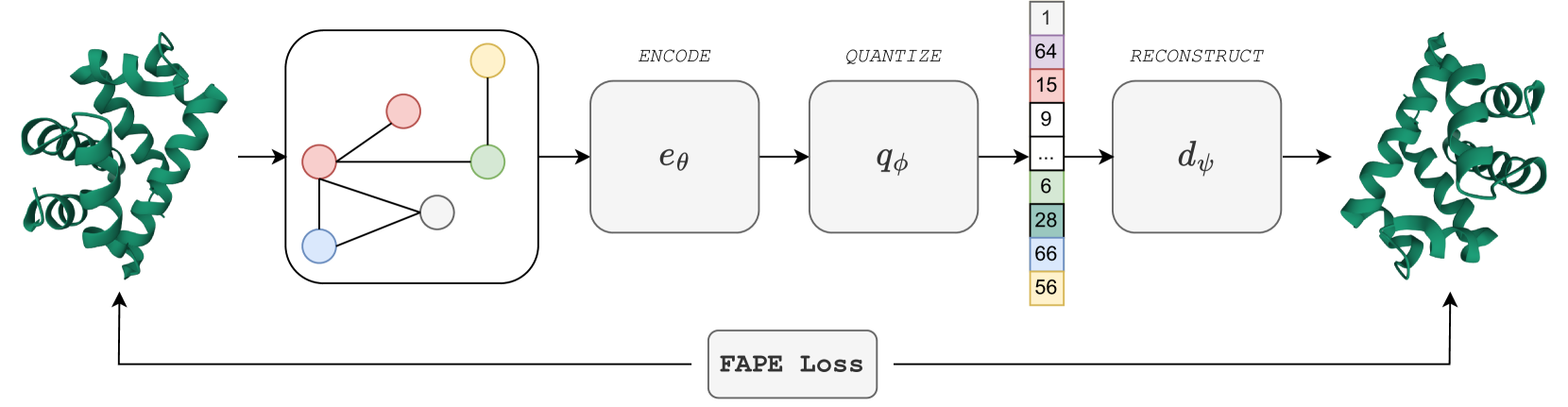
Representation learning and emph{de novo} generation of proteins are pivotal computational biology tasks. Whilst natural language processing (NLP) techniques have proven highly effective for protein sequence modelling, structure modelling presents a complex challenge, primarily due to its continuous and three-dimensional nature. Motivated by this discrepancy, we introduce an approach using a vector-quantized autoencoder that effectively tokenizes protein structures into discrete representations. This method transforms the continuous, complex space of protein structures into a manageable, discrete format with a codebook ranging from 4096 to 64000 tokens, achieving high-fidelity reconstructions with backbone root mean square deviations (RMSD) of approximately 1-5 AA. To demonstrate the efficacy of our learned representations, we show that a simple GPT model trained on our codebooks can generate novel, diverse, and designable protein structures. Our approach not only provides representations of protein structure, but also mitigates the challenges of disparate modal representations and sets a foundation for seamless, multi-modal integration, enhancing the capabilities of computational methods in protein design.
Read more5/28/2024
0
Protein Representation Learning by Capturing Protein Sequence-Structure-Function Relationship
Eunji Ko, Seul Lee, Minseon Kim, Dongki Kim
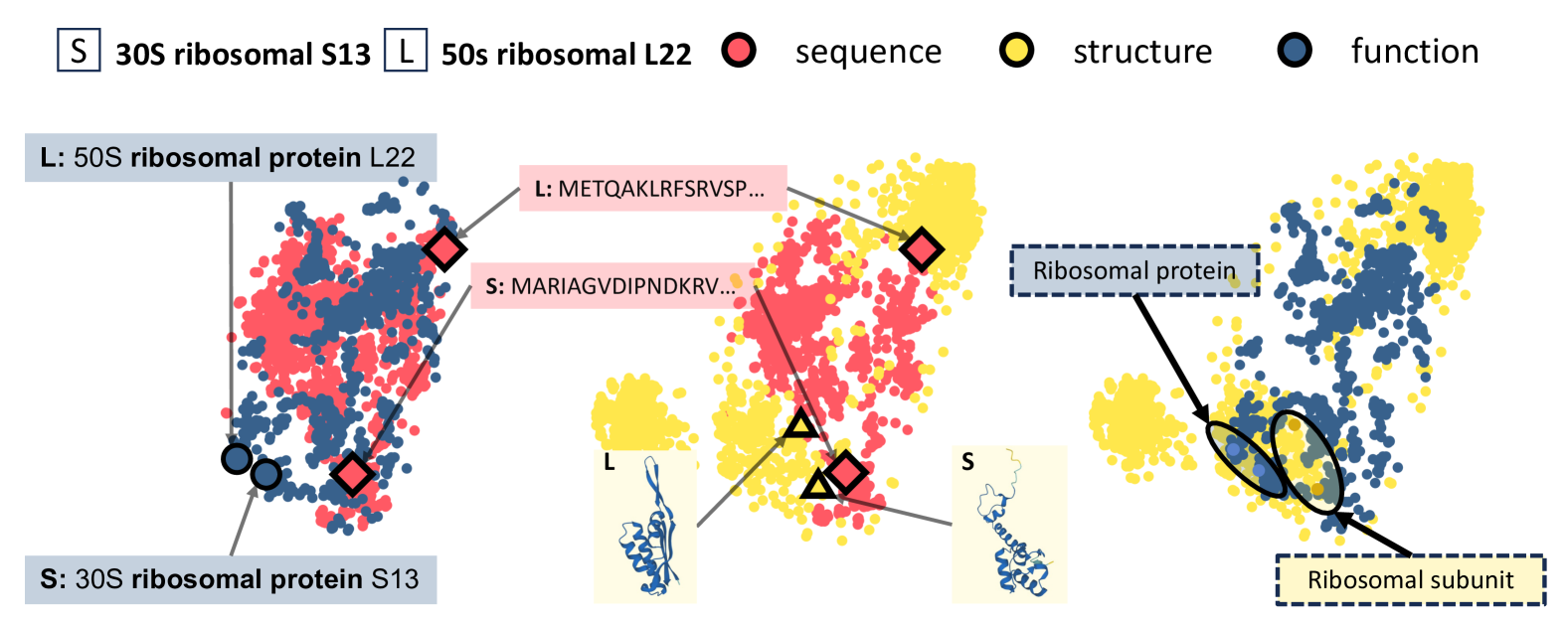
The goal of protein representation learning is to extract knowledge from protein databases that can be applied to various protein-related downstream tasks. Although protein sequence, structure, and function are the three key modalities for a comprehensive understanding of proteins, existing methods for protein representation learning have utilized only one or two of these modalities due to the difficulty of capturing the asymmetric interrelationships between them. To account for this asymmetry, we introduce our novel asymmetric multi-modal masked autoencoder (AMMA). AMMA adopts (1) a unified multi-modal encoder to integrate all three modalities into a unified representation space and (2) asymmetric decoders to ensure that sequence latent features reflect structural and functional information. The experiments demonstrate that the proposed AMMA is highly effective in learning protein representations that exhibit well-aligned inter-modal relationships, which in turn makes it effective for various downstream protein-related tasks.
Read more5/14/2024
0
Contrastive learning of T cell receptor representations
Yuta Nagano, Andrew Pyo, Martina Milighetti, James Henderson, John Shawe-Taylor, Benny Chain, Andreas Tiffeau-Mayer
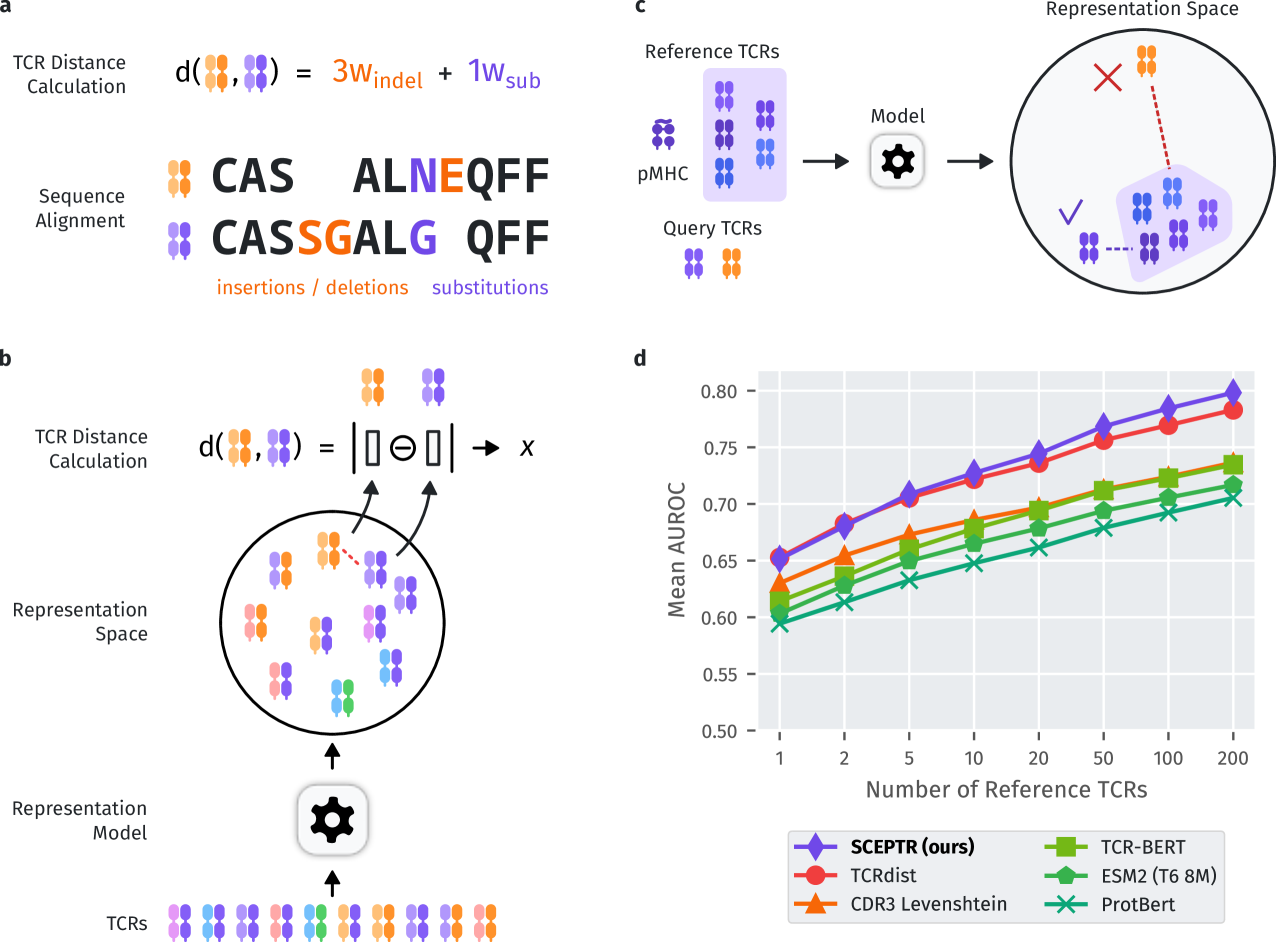
Computational prediction of the interaction of T cell receptors (TCRs) and their ligands is a grand challenge in immunology. Despite advances in high-throughput assays, specificity-labelled TCR data remains sparse. In other domains, the pre-training of language models on unlabelled data has been successfully used to address data bottlenecks. However, it is unclear how to best pre-train protein language models for TCR specificity prediction. Here we introduce a TCR language model called SCEPTR (Simple Contrastive Embedding of the Primary sequence of T cell Receptors), capable of data-efficient transfer learning. Through our model, we introduce a novel pre-training strategy combining autocontrastive learning and masked-language modelling, which enables SCEPTR to achieve its state-of-the-art performance. In contrast, existing protein language models and a variant of SCEPTR pre-trained without autocontrastive learning are outperformed by sequence alignment-based methods. We anticipate that contrastive learning will be a useful paradigm to decode the rules of TCR specificity.
Read more6/11/2024