Enhanced Gene Selection in Single-Cell Genomics: Pre-Filtering Synergy and Reinforced Optimization

0
🛠️
Sign in to get full access
Overview
- The paper introduces an iterative gene panel selection strategy for clustering tasks in single-cell genomics.
- The method aims to address the limitations of traditional gene selection approaches, which can be prone to biases and inefficiencies.
- The proposed approach integrates results from other gene selection algorithms and leverages reinforcement learning to refine the gene panel selection dynamically.
Plain English Explanation
Single-cell genomics is a rapidly advancing field that allows scientists to analyze the genetic makeup of individual cells. This technique is important for understanding complex biological systems. However, the vast amount of data generated by single-cell analysis can be challenging to interpret effectively.
The key to unlocking insights from this data lies in selecting the most informative genes - the ones that contribute the most to the specific analysis task at hand. Traditional gene selection methods often rely on expert knowledge or heuristic-based approaches, which can sometimes miss critical genomic signals.
To address these limitations, the researchers in this study have developed a new strategy for gene panel selection. Their method integrates the results from various gene selection algorithms, using them as a starting point to guide the search for the optimal gene panel. The researchers then incorporate the principles of reinforcement learning, which allows the system to continuously refine and optimize the gene panel selection based on the feedback it receives.
This combination of different approaches helps to mitigate the biases that can be present in the initial gene selection and enables the system to dynamically adapt to the specific needs of the analysis task. By focusing on the most informative genes, the researchers aim to streamline the interpretation of complex single-cell genomic data and uncover critical insights.
Technical Explanation
The researchers introduce an iterative gene panel selection strategy for single-cell genomic clustering tasks. Their method leverages the strengths of various gene selection algorithms, using their results as initial boundaries or prior knowledge to guide the search for the optimal gene panel.
The researchers recognize the limitations of traditional gene selection methods, which often rely on expert domain knowledge, embedded machine learning models, or heuristic-based iterative optimization. These approaches can be prone to biases and inefficiencies, potentially obscuring critical genomic signals.
To address these challenges, the researchers incorporate the stochastic nature of the exploration process in reinforcement learning (RL) and its capability for continuous optimization through reward-based feedback. This combination helps to mitigate the biases inherent in the initial boundaries and allows the system to refine and target gene panel selection dynamically.
The researchers conducted detailed comparative experiments, case studies, and visualization analysis to demonstrate the effectiveness of their method. By integrating the results from other gene selection algorithms and leveraging the adaptability of reinforcement learning, the researchers aim to transcend the constraints of traditional approaches and enable more efficient and informed analysis of single-cell genomic data.
Critical Analysis
The researchers have proposed a novel and promising approach to gene panel selection for single-cell genomic analysis. By integrating the results from various gene selection algorithms and incorporating reinforcement learning principles, the method aims to address the limitations of traditional approaches, which can be prone to biases and inefficiencies.
One potential area for further research could be the exploration of different reinforcement learning algorithms or reward functions to further optimize the gene panel selection process. The researchers mention the stochastic nature of the exploration process in RL, but additional investigation into the impact of different RL techniques could yield valuable insights.
Additionally, while the researchers have conducted comprehensive experiments and case studies, it would be interesting to see how their method performs on a wider range of single-cell genomic datasets and analysis tasks. Exploring the scalability and generalizability of the approach could strengthen the claims about its effectiveness.
Overall, the researchers have presented a thoughtful and innovative approach to gene panel selection in single-cell genomics. By leveraging the strengths of multiple gene selection algorithms and the adaptability of reinforcement learning, the proposed method offers a promising step forward in streamlining the analysis of complex single-cell genomic data.
Conclusion
The researchers in this study have introduced an iterative gene panel selection strategy for single-cell genomic clustering tasks. By integrating the results from various gene selection algorithms and incorporating the principles of reinforcement learning, the proposed method aims to address the limitations of traditional approaches, which can be prone to biases and inefficiencies.
The integration of multiple gene selection algorithms and the dynamic optimization enabled by reinforcement learning offer a refined and versatile approach to identifying the most informative genes for single-cell genomic analysis. This strategy has the potential to enhance the interpretation of complex biological data and uncover critical insights that may have been obscured by the biases inherent in traditional gene selection methods.
As the field of single-cell genomics continues to evolve, the researchers' innovative approach to gene panel selection could have far-reaching implications for researchers and clinicians working to understand the intricacies of complex biological systems.
This summary was produced with help from an AI and may contain inaccuracies - check out the links to read the original source documents!
Related Papers
🛠️
0
Enhanced Gene Selection in Single-Cell Genomics: Pre-Filtering Synergy and Reinforced Optimization
Weiliang Zhang, Zhen Meng, Dongjie Wang, Min Wu, Kunpeng Liu, Yuanchun Zhou, Meng Xiao
Recent advancements in single-cell genomics necessitate precision in gene panel selection to interpret complex biological data effectively. Those methods aim to streamline the analysis of scRNA-seq data by focusing on the most informative genes that contribute significantly to the specific analysis task. Traditional selection methods, which often rely on expert domain knowledge, embedded machine learning models, or heuristic-based iterative optimization, are prone to biases and inefficiencies that may obscure critical genomic signals. Recognizing the limitations of traditional methods, we aim to transcend these constraints with a refined strategy. In this study, we introduce an iterative gene panel selection strategy that is applicable to clustering tasks in single-cell genomics. Our method uniquely integrates results from other gene selection algorithms, providing valuable preliminary boundaries or prior knowledge as initial guides in the search space to enhance the efficiency of our framework. Furthermore, we incorporate the stochastic nature of the exploration process in reinforcement learning (RL) and its capability for continuous optimization through reward-based feedback. This combination mitigates the biases inherent in the initial boundaries and harnesses RL's adaptability to refine and target gene panel selection dynamically. To illustrate the effectiveness of our method, we conducted detailed comparative experiments, case studies, and visualization analysis.
Read more6/12/2024
✨
0
Quantum Annealing for Enhanced Feature Selection in Single-Cell RNA Sequencing Data Analysis
Selim Romero, Shreyan Gupta, Victoria Gatlin, Robert S. Chapkin, James J. Cai
Feature selection is vital for identifying relevant variables in classification and regression models, especially in single-cell RNA sequencing (scRNA-seq) data analysis. Traditional methods like LASSO often struggle with the nonlinearities and multicollinearities in scRNA-seq data due to complex gene expression and extensive gene interactions. Quantum annealing, a form of quantum computing, offers a promising solution. In this study, we apply quantum annealing-empowered quadratic unconstrained binary optimization (QUBO) for feature selection in scRNA-seq data. Using data from a human cell differentiation system, we show that QUBO identifies genes with nonlinear expression patterns related to differentiation time, many of which play roles in the differentiation process. In contrast, LASSO tends to select genes with more linear expression changes. Our findings suggest that the QUBO method, powered by quantum annealing, can reveal complex gene expression patterns that traditional methods might overlook, enhancing scRNA-seq data analysis and interpretation.
Read more8/30/2024
🤿
0
Pan-cancer gene set discovery via scRNA-seq for optimal deep learning based downstream tasks
Jong Hyun Kim, Jongseong Jang
The application of machine learning to transcriptomics data has led to significant advances in cancer research. However, the high dimensionality and complexity of RNA sequencing (RNA-seq) data pose significant challenges in pan-cancer studies. This study hypothesizes that gene sets derived from single-cell RNA sequencing (scRNA-seq) data will outperform those selected using bulk RNA-seq in pan-cancer downstream tasks. We analyzed scRNA-seq data from 181 tumor biopsies across 13 cancer types. High-dimensional weighted gene co-expression network analysis (hdWGCNA) was performed to identify relevant gene sets, which were further refined using XGBoost for feature selection. These gene sets were applied to downstream tasks using TCGA pan-cancer RNA-seq data and compared to six reference gene sets and oncogenes from OncoKB evaluated with deep learning models, including multilayer perceptrons (MLPs) and graph neural networks (GNNs). The XGBoost-refined hdWGCNA gene set demonstrated higher performance in most tasks, including tumor mutation burden assessment, microsatellite instability classification, mutation prediction, cancer subtyping, and grading. In particular, genes such as DPM1, BAD, and FKBP4 emerged as important pan-cancer biomarkers, with DPM1 consistently significant across tasks. This study presents a robust approach for feature selection in cancer genomics by integrating scRNA-seq data and advanced analysis techniques, offering a promising avenue for improving predictive accuracy in cancer research.
Read more8/15/2024
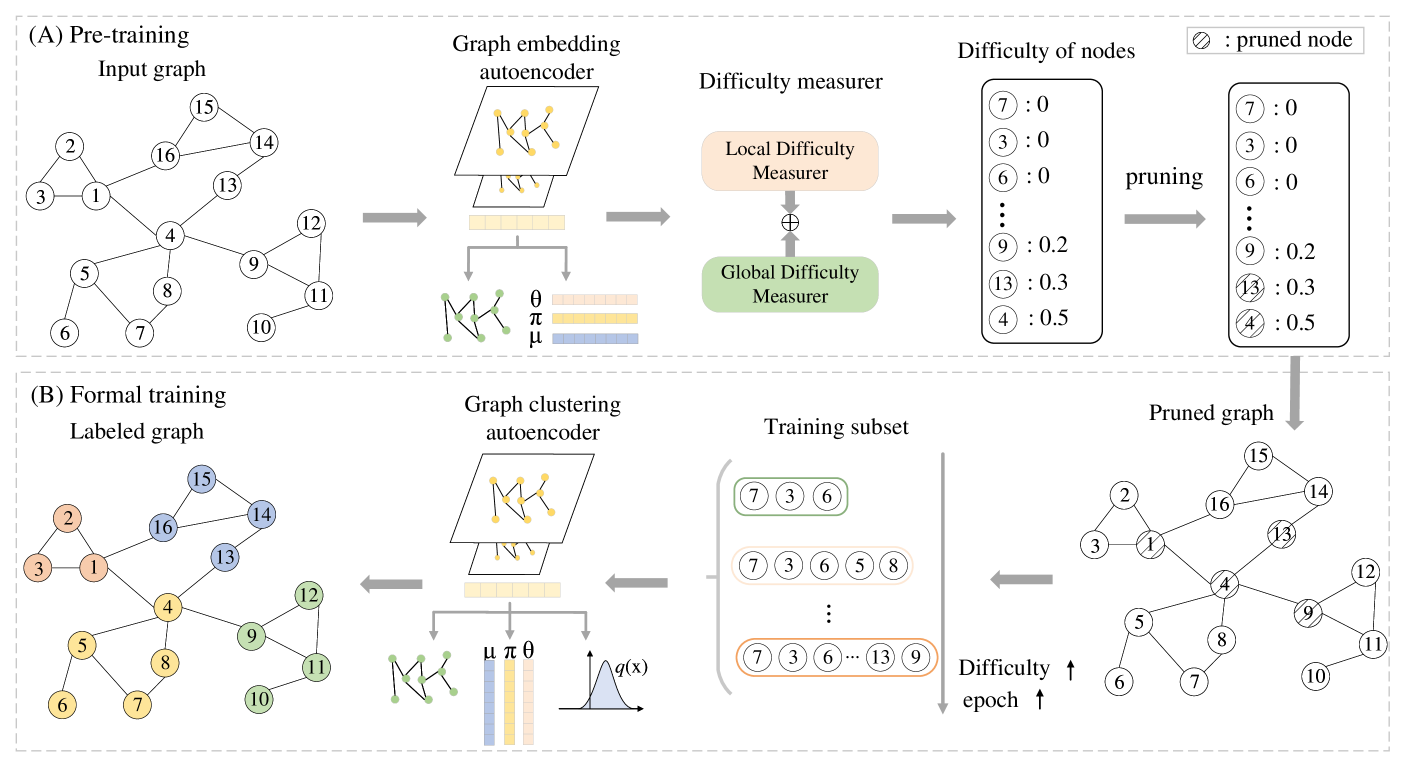
0
Single-cell Curriculum Learning-based Deep Graph Embedding Clustering
Huifa Li, Jie Fu, Xinpeng Ling, Zhiyu Sun, Kuncan Wang, Zhili Chen
The swift advancement of single-cell RNA sequencing (scRNA-seq) technologies enables the investigation of cellular-level tissue heterogeneity. Cell annotation significantly contributes to the extensive downstream analysis of scRNA-seq data. However, The analysis of scRNA-seq for biological inference presents challenges owing to its intricate and indeterminate data distribution, characterized by a substantial volume and a high frequency of dropout events. Furthermore, the quality of training samples varies greatly, and the performance of the popular scRNA-seq data clustering solution GNN could be harmed by two types of low-quality training nodes: 1) nodes on the boundary; 2) nodes that contribute little additional information to the graph. To address these problems, we propose a single-cell curriculum learning-based deep graph embedding clustering (scCLG). We first propose a Chebyshev graph convolutional autoencoder with multi-decoder (ChebAE) that combines three optimization objectives corresponding to three decoders, including topology reconstruction loss of cell graphs, zero-inflated negative binomial (ZINB) loss, and clustering loss, to learn cell-cell topology representation. Meanwhile, we employ a selective training strategy to train GNN based on the features and entropy of nodes and prune the difficult nodes based on the difficulty scores to keep the high-quality graph. Empirical results on a variety of gene expression datasets show that our model outperforms state-of-the-art methods.
Read more8/21/2024